La scoperta e lo sviluppo di nuovi farmaci rappresentano uno dei campi più dinamici e rigorosi della scienza, un processo che dall'antica estrazione di sostanze naturali si è evoluto in un'impresa multidisciplinare e tecnologicamente avanzata. Questo percorso non solo mira a trovare nuove soluzioni terapeutiche, ma anche a garantirne l'efficacia e la sicurezza per milioni di persone in tutto il mondo.
Il Concetto di Farmaco: Una Definizione Storica e Moderna
Il termine "farmaco" deriva dal greco "pharmacon", che già nel 460 a.C. Ippocrate, considerato il padre della medicina, definì come “un preparato capace di determinare un'azione sull'organismo, modificandone lo stato esistente”. Questa definizione, seppur antica, conserva la sua essenza, evidenziando la duplice natura di una sostanza che può sia curare che essere nociva. Oggi, un farmaco è composto dal principio attivo, responsabile dell’effetto curativo, e da altre sostanze, chiamate eccipienti, che non hanno proprietà terapeutiche ma facilitano la produzione del farmaco e ne migliorano le prestazioni dall'assunzione all'assorbimento.
L'Evoluzione della Scoperta dei Principi Attivi
Nei tempi antichi, i principi attivi per la medicina venivano solitamente estratti da piante o animali. Anche se alcuni ingredienti specifici provengono ancora da una fonte biologica, i nuovi prodotti farmaceutici raramente vengono più ottenuti attraverso questi metodi, semplicemente perché, grazie a secoli di storia, sono già stati tutti inventati. Storicamente, quando veniva scoperto un nuovo farmaco, non si sapeva come avrebbe interagito con il corpo e il suo scopo come farmaco; questo è stato trovato solo dopo la scoperta del farmaco.
Con i progressi scientifici e le nostre attuali conoscenze, tuttavia, è diventato più tipico eseguire questo processo inverso chiamato “scoperta di farmaci basata su target”. Oggi, la ricerca farmacologica comincia con la scoperta di un composto dotato di "attività farmacologica", ovvero con la capacità di modificare un processo biologico.
Sintesi Chimica e Biotecnologia
La sintesi chimica è ciò che ha plasmato l’industria farmaceutica come la conosciamo. Un esempio di reazione chimica che potresti conoscere è il modo in cui si forma il calcestruzzo: quando si mescola la polvere di cemento con acqua e alcuni altri ingredienti, avviene un processo chimico e si trasforma in cemento con proprietà chimiche diverse. Questo processo non è reversibile; se si macinasse il calcestruzzo, non si potrebbe semplicemente aggiungere nuovamente acqua per usarlo come cemento; è già diventato qualcos'altro.
Per quanto riguarda vaccini e antibiotici, l’industria farmaceutica utilizza prevalentemente la biotecnologia anziché la sintesi chimica. In genere il corpo avrebbe sostanze per combattere le malattie per natura. Ma se mancano o non sono sufficientemente presenti nel corpo, si dispone di microrganismi modificati che possono creare quelle sostanze nel corpo utilizzando una tecnologia intelligente.
L'Identificazione del Bersaglio Farmacologico
Per sviluppare un buon farmaco è necessario dapprima individuare il giusto bersaglio da colpire. Generalmente si punta sulle proteine, elementi particolarmente importanti all’interno di specifici percorsi biologici, chiamati “pathway”. Un buon farmaco deve essere in grado di legarsi a una proteina in modo analogo ad una chiave che entra in una serratura. Se la chiave è il farmaco, la serratura sarà la proteina in grado di attivarsi e dare il via alla risposta terapeutica.

Così come tra le chiavi ci sono quelle che entrano perfettamente nelle serrature e quelle che, invece, entrano e si bloccano, così tra i farmaci esistono quelli che si legano ad una specifica molecola, causando una risposta (agonisti), e quelli che invece lo bloccano soltanto (antagonisti). Il "bersaglio farmacologico" è l'elemento o il meccanismo biologico sul quale il farmaco dovrà intervenire per modificare l'evolversi di una malattia. A seconda della patologia, il bersaglio può essere diverso: un virus o un batterio, la carenza di un ormone nelle patologie metaboliche, il meccanismo di degenerazione delle cellule cerebrali come nella malattia di Parkinson o nel morbo di Alzheimer. Quindi il bersaglio può essere una proteina difettosa, un microrganismo, un segnale biochimico malfunzionante, un legame molecolare alterato.
La Ricerca del Principio Attivo
Una volta individuato il bersaglio farmacologico, si procede con l'identificazione di sostanze in grado di legarsi ad esso in modo specifico e selettivo così da ottenere un effetto terapeutico. Queste sostanze sono i principi attivi e rappresentano i precursori del futuro farmaco, chiamati anche composti guida (lead compound).
Per trovare il principio attivo si possono seguire diversi approcci:
- Approccio empirico ("screening"): Si basa sullo "screening" di composti già esistenti. Si analizzano vere e proprie librerie di molecole chimiche, allo scopo di individuare quella giusta come punto di partenza per costruire un nuovo farmaco.
- Approccio moderno ("rational drug design"): Presuppone la conoscenza fine dei meccanismi di una malattia. In questo caso il composto viene progettato “a tavolino”. Fino ad ora questo secondo metodo ha generato pochi frutti rispetto alla grande massa di medicine trovate nel secolo scorso con il metodo empirico: la selezione di molecole chimiche più efficaci per un problema e meno tossiche, però, lascia in secondo piano le domande sui meccanismi d'azione.
- Scoperta casuale: In alcuni casi, l'identificazione delle molecole guida avviene casualmente, come nel caso della penicillina, il primo antibiotico scoperto, identificato da Fleming perché una coltura di Staphylococcus aureus era stata inquinata da una muffa. In altri casi, la scoperta avviene osservando gli effetti secondari positivi di una molecola di cui si cercava di individuare gli effetti indesiderati.
Gli scienziati possono comprendere la causa di una malattia e quali sono le cellule e i loro recettori coinvolti. Una volta che una molecola recettore target o un enzima sono stati individuati, gli scienziati iniziano a cercare eventuali composti che interagiscano con il target al fine di correggere l'attività della malattia. Tale ricerca può comprendere "librerie" di milioni di molecole sviluppate da aziende farmaceutiche. Le molecole scoperte più promettenti saranno quindi modificate in diversi modi allo scopo di provare a produrre un farmaco efficace con pochi effetti collaterali negativi.
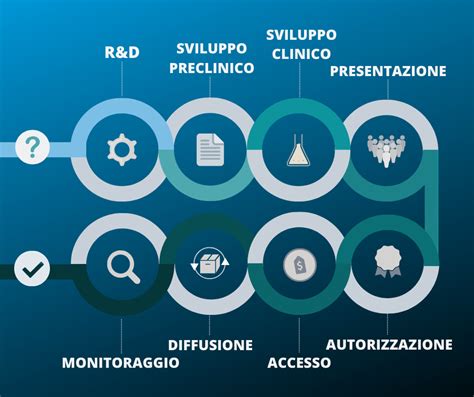
Le Fasi dello Sviluppo di un Farmaco: Un Percorso Rigoroso
Lo sviluppo di nuovi farmaci è un processo che segue tappe molto definite. Dalla prima idea di un nuovo trattamento alla sua commercializzazione passano in media dai 10 ai 12 anni, ma il percorso può durare anche molto di più. È un lungo percorso segnato, a volte, da qualche insuccesso ma, soprattutto, dal raggiungimento di traguardi che hanno consentito di debellare gravi malattie e di migliorare la durata e la qualità della vita dell’uomo.
Drug discovery...alla scoperta di un farmaco
Fase Preclinica: Il Laboratorio e gli Animali
Dopo aver individuato il bersaglio farmacologico e aver scelto il principio attivo più giusto, il ricercatore deve accertare la reale efficacia del composto candidato prima in modelli sperimentali. Questi studi fanno parte della fase preclinica dello sviluppo di un farmaco, fase che precede le prove cliniche del farmaco nei pazienti e che richiede da 2 a 3 anni e che costituisce circa il 30% dell’investimento economico totale.
- Studi in vitro: I primi test si effettuano in laboratorio, su cellule di laboratorio (modelli in vitro) o in provetta. Lo scopo è valutare per la prima volta la sicurezza, la tossicità e la possibile attività della molecola. La sostanza viene messa in provetta insieme a colture cellulari o a microrganismi e sottoposta a una serie di test. Questi esperimenti vengono eseguiti in laboratori altamente specializzati.
- Studi in vivo: Soltanto quando si è appurato in laboratorio che la molecola possiede potenziali effetti terapeutici si può passare alla sperimentazione sugli animali (studi “in vivo”). Tradizionalmente, gran parte di questa ricerca veniva eseguita su animali, ma grazie al progresso tecnologico e alle nostre conoscenze scientifiche, gran parte dei test può essere eseguita anche su colture cellulari in piastre Petri. L’utilizzo di animali di laboratorio per la sperimentazione sui nuovi farmaci, pur essendo necessario per legge e regolamentato da norme giuridiche severe, è un tema controverso. Tuttavia, la complessità dell’organismo umano è impossibile da ricostruire soltanto in simulazioni al computer, in cellule in coltura, isolate dagli altri organi, e persino con ricostruzioni artificiali degli organi, i cosiddetti organoidi. Con animali come i topi, invece, abbiamo in comune buona parte del DNA e molti tessuti e organi somigliano fortemente ai nostri. I risultati ottenuti con la sperimentazione animale offrono indicazioni importanti sulla tollerabilità e l’efficacia dei farmaci, che per ora non si possono ottenere altrimenti. Proprio per queste ragioni la sperimentazione clinica dei farmaci in animali di laboratorio di diverse specie è obbligatoria per legge.
I test di laboratorio (preclinici) devono analizzare la tossicità, la farmacocinetica e la farmacodinamica.
- Tossicologia: Si dimostra che l’effetto terapeutico è conseguenza del processo biologico colpito dal farmaco e, contemporaneamente, che non si accompagni a eventi sfavorevoli su altri organi o funzioni. Gli studi di tossicità acuta e cronica controllano se il farmaco impatta sulla capacità riproduttiva umana e sulla cancerogenesi.
- Farmacocinetica: È quella branca della farmacologia che studia il comportamento dei farmaci introdotti nell'organismo: l'assorbimento; la distribuzione negli organi, nei tessuti e nelle cellule; le trasformazioni metaboliche; le vie e le modalità di eliminazione. Questi studi forniscono indicazioni sulla sede e sul meccanismo d'azione dei medicinali, sulla rapidità e sulla durata dei loro effetti, su eventuali fenomeni di accumulo e su altri fattori la cui conoscenza è necessaria affinché possa essere stabilita una presunta dose da somministrare all'uomo.
- Farmacodinamica: Si valuta la risposta dei tessuti e delle cellule al farmaco.
Dall'insieme dei risultati di tossicità, farmacocinetica e farmacodinamica si ricava l’indice terapeutico, cioè l’indice della sicurezza di un farmaco calcolato dal rapporto tra l’efficacia e la tossicità di un prodotto. Idealmente ogni buon farmaco dovrebbe avere un alto indice terapeutico, e cioè dovrebbe essere molto efficace e tollerabile e poco dannoso.
Mentre si completa la fase sperimentale, si iniziano studi anche piuttosto complessi di tecnica farmaceutica: se il prodotto va somministrato per via orale è meglio in pastiglie, compresse, capsule o ovuli? E poi: a rilascio lento o rapido? Il principio attivo, infatti, deve essere stabile per lungo tempo, non deve alterarsi e deve resistere ai cambiamenti di temperatura. Tutto ciò grazie all'aggiunta di eventuali eccipienti.
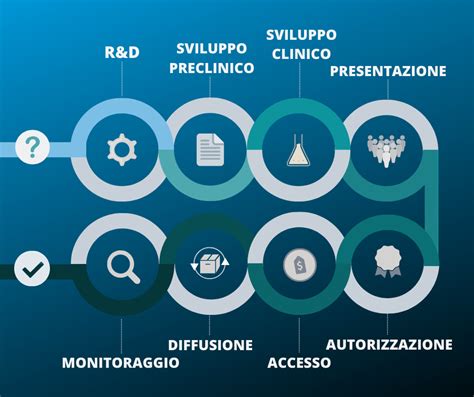
Sperimentazione Clinica: Le Fasi sull'Uomo
Una volta superata la fase preclinica, la molecola valutata come più promettente tra quelle prese in considerazione viene avviata alla cosiddetta sperimentazione clinica, negli esseri umani. Questo percorso coinvolge volontari sani oppure pazienti critici. In corso di studio clinico le caratteristiche del nuovo farmaco passate al vaglio sono: qualità, sicurezza ed efficacia.
Il promotore (la casa farmaceutica, l'azienda biotecnologica, il medico o il centro pubblico che ha scoperto la molecola) presenta i risultati dei test pre-clinici all'AIFA (Agenzia Italiana del Farmaco) per ottenere l'autorizzazione a uno studio clinico. Una volta autorizzata la sperimentazione umana, si procede attraverso tre fasi principali.
Fase 1: Sicurezza e Tollerabilità
Questa fase ha lo scopo di fornire una prima valutazione della sicurezza e tollerabilità del medicinale. In genere, questi studi sono condotti in pochi centri selezionati, su un numero limitato (20-100) di volontari sani, in età non avanzata, per i quali è documentata l’assenza e valutata la non predisposizione a malattie. L'obiettivo principale è la valutazione degli effetti collaterali che possono essere attesi considerando i risultati delle precedenti sperimentazioni sugli animali e la valutazione della modalità di azione e distribuzione del farmaco nell’organismo (farmacocinetica e farmacodinamica). I volontari vengono divisi in più gruppi, ciascuno dei quali riceve una diversa dose di farmaco (in genere crescente), per valutare gli eventuali effetti indesiderati della sostanza in relazione alla quantità somministrata. Se oggetto della sperimentazione sono gravi patologie (per esempio tumori, AIDS), questi studi possono essere condotti direttamente su pazienti che ne sono affetti e per i quali il farmaco è stato pensato. Se il farmaco dimostra di avere un livello di tossicità accettabile rispetto al beneficio previsto (profilo beneficio/rischio) allora può passare alle successive fasi della sperimentazione. Questa fase è cruciale per stabilire la dose che garantisca il giusto effetto biologico senza essere tossica.
Fase 2: Efficacia Preliminare e Dosaggio Ottimale
Nello studio di fase 2 (definito anche terapeutico-esplorativo) comincia ad essere indagata l’attività terapeutica del potenziale farmaco, cioè la sua capacità di produrre sull’organismo umano gli effetti curativi desiderati. Questa fase serve inoltre a comprendere quale sarà la dose migliore da sperimentare nelle fasi successive, e determinare l’effetto del farmaco in relazione ad alcuni parametri (come, ad esempio, la pressione sanguigna) considerati indicatori della salute del paziente. Negli studi di fase 2 la sostanza è somministrata a soggetti volontari affetti dalla patologia per cui il farmaco è stato pensato. I soggetti “arruolati” per lo studio vengono generalmente divisi in più gruppi (da 100 a 300 pazienti), a ciascuno dei quali è somministrata una dose differente del farmaco e, quando è eticamente possibile, un placebo (vale a dire una sostanza priva di efficacia terapeutica). Per evitare che la somministrazione del placebo influenzi le aspettative dei partecipanti, le valutazioni dei parametri di attività e sicurezza sono condotte senza che paziente (studio in cieco singolo), o medico e paziente (studio in doppio cieco), conoscano il tipo di trattamento ricevuto o somministrato. Questa fase dura circa un paio d'anni. Questa seconda fase è utile quindi a dimostrare la non tossicità e l’attività del nuovo principio attivo sperimentale.
Fase 3: Efficacia Confermata e Confronto con Standard
A tutte le domande su efficacia, benefici rispetto a farmaci simili e rapporto rischio-beneficio si risponde con lo studio di fase 3 (o terapeutico-confermatorio). In questo caso non sono più poche decine i pazienti “arruolati”, ma centinaia o migliaia (fino a più di 5000 volontari), spesso in diverse nazioni. L’efficacia del farmaco sui sintomi, sulla qualità della vita o sulla sopravvivenza è confrontata con un placebo (sostanza priva di efficacia terapeutica), con altri farmaci già in uso, o con nessun trattamento. La tipologia di studio di riferimento in questa fase è lo Studio clinico controllato randomizzato. Si tratta di un tipo di studio in cui ai pazienti viene assegnato casualmente (in inglese random) il nuovo principio attivo o un farmaco di controllo (in genere il trattamento standard per quella specifica patologia oggetto della ricerca). Questo garantisce che i due gruppi siano simili per tutte le caratteristiche salvo che per il medicinale assunto. Dunque, alla fine della sperimentazione, sarà possibile attribuire ogni differenza nella salute dei partecipanti esclusivamente al trattamento e non a errori o al caso. Durante questa fase vengono controllate con molta attenzione l'insorgenza, la frequenza e gravità degli effetti indesiderati. La durata della somministrazione del farmaco è variabile a seconda degli obiettivi che la sperimentazione si pone, ma in genere dura dei mesi. Il periodo di monitoraggio degli effetti del farmaco è invece spesso più lungo, arrivando in qualche caso a 3-5 anni.
Il risultato della fase 3 è una risposta in grado di valutare il rapporto rischio-beneficio. È importante che nei due gruppi a confronto i pazienti vengano collocati “a caso”, in gergo si dice “randomizzati”, e “in doppio cieco”, cioè né il paziente né il medico curante devono essere a conoscenza di quale medicinale viene somministrato, per evitare che aspettative o altri fattori possano influenzare il giudizio sull'efficacia e sugli effetti collaterali del farmaco in esame.
Purtroppo, non fila tutto sempre liscio: in ogni momento di questa delicata sequenza di studi è possibile che si debba ricominciare dall'inizio con lo stesso principio attivo o con altre sostanze chimiche.
Il Ruolo dei Comitati Etici e dei Protocolli
La ricerca su nuovi farmaci segue una serie di tappe codificate a livello internazionale, alle quali i ricercatori devono attenersi. Si tratta di un meccanismo messo in piedi nel tempo proprio a tutela della salute dei malati. Oltre a consentire risultati affidabili, le regole delle sperimentazioni danno notevole importanza anche ai diritti, alla sicurezza e al benessere delle persone che prendono parte alle sperimentazioni. L’insieme dei principi che devono guidare la ricerca è regolato dall’Unione Europea, con le norme di Good Clinical Practice (in italiano, di buona pratica clinica).
Il presupposto è che nessun farmaco, neanche il più preciso e mirato, è mai privo di effetti collaterali. Ogni composto può provocare dei danni, grandi o piccoli, che devono essere individuati e compresi prima che la cura sia approvata e messa a disposizione di tutti i pazienti. Per questo motivo, la ricerca clinica su ogni nuova molecola parte dall’elaborazione di un protocollo in cui si stabilisce, tra le altre cose, quali caratteristiche dovranno avere le persone per entrare nella sperimentazione (per esempio, sesso, fascia d’età, tipo di malattia, gravità e così via) affinché i risultati siano i più chiari possibili.
Il protocollo sperimentale deve essere sottoposto per approvazione a una serie di enti di controllo sia scientifici, sia etici. In Italia la valutazione coinvolge l’Agenzia Italiana del Farmaco (AIFA), l’Istituto Superiore di Sanità e appositi comitati etici. Il comitato etico dell'ospedale e il consenso informato del paziente sono necessari. Il farmaco deve essere stato inserito preventivamente in un elenco, presso l’Agenzia Italiana del Farmaco (AIFA), e deve avere superato la fase III di sperimentazione. Un protocollo descrive tempi e dosi di somministrazione della molecola, numero e frequenza di visite di controllo, tipo di analisi da eseguire a ogni visita e ogni altro dettaglio tecnico dello studio: qualsiasi cosa andrà fatta al paziente, deve essere specificata nel protocollo nei minimi dettagli.
Un consenso informato è un documento con valore legale attraverso cui s'informa il paziente tutti i passaggi che saranno seguiti durante lo studio, rischi, benefici (potenziali), la durata dello studio, il suo scopo, eccetera; in altre parole, è il documento con la cui firma il paziente autorizza il centro di ricerca ad effettuare lo studio sulla sua persona. Entrambi questi documenti dovranno passare al vaglio dal comitato etico dell'Agenzia del Farmaco, da quello di ogni singolo centro di ricerca incluso nello studio e, in molti casi, da quello del promotore stesso.
Fase 4: Farmacovigilanza e Monitoraggio Post-Marketing
Una volta che il nuovo trattamento è a disposizione di tutti, i controlli non vengono meno. La cosiddetta fase IV, di farmacovigilanza, chiamata anche sorveglianza post-marketing, serve a raccogliere segnalazioni di tutti gli effetti collaterali, tra cui quelli molto rari, dell’ordine di un caso su milioni di utilizzatori, che non sono comparsi nelle precedenti fasi di sperimentazione. Qualsiasi effetto collaterale, sia pur minimo e non notato nelle fasi precedenti, viene segnalato alle autorità, che ne valutano l’importanza e possono cambiare le indicazioni o integrare il foglietto illustrativo. In casi estremi, tali autorità possono disporre il ritiro dal commercio del farmaco.
Il farmaco continua ad essere monitorato per verificarne la tollerabilità e la sicurezza a lungo termine. È quando un farmaco si trova in questa fase che si potrebbe leggere la notizia "Farmaco ritirato dal commercio": si tratta di casi estremamente rari, dato che il più delle volte si ritirano semplicemente dei lotti che, nonostante tutti i rigorosissimi controlli di qualità, si sono rivelati difettosi. Nonostante ciò sono comunque casi gravi e da tenere in conto.
Chi Sviluppa e Chi Approva un Nuovo Farmaco?
Non esiste una sola figura addetta allo sviluppo di un nuovo medicinale. Per creare nuovi farmaci sono necessarie molte competenze diverse: servono chirurghi e patologi che conoscono bene le malattie dell’uomo e le loro manifestazioni, servono biologi che non si perdono fra i tanti processi e le tante molecole del nostro corpo, servono chimici e fisici che conoscono le leggi della materia e la sappiano manipolare, servono farmacologi che conoscono le migliaia di farmaci di ieri e di oggi comprese proprietà ed effetti, servono veterinari per sviluppare modelli di laboratorio per studiare malattie umane, servono ingegneri in grado di inventare macchine piccole e grandi per incapsulare e trasportare i farmaci, servono matematici e statistici per interpretare i numeri di esperimenti e studi clinici, servono informatici per organizzare tutte le informazioni e simulare l'incontro fra farmaco e organismo prima di trasferire la simulazione alla realtà clinica, servono manager della ricerca in grado di gestire la complessità organizzativa dello sviluppo dei farmaci e della loro sperimentazione.
Periodicamente nuovi farmaci vengono immessi sul mercato e altri invece vengono ritirati. Ma chi è che dirige queste operazioni, dietro le quinte? Le decisioni vengono prese in sede europea dall'EMA - European Medicines Agency. L'autorizzazione di un nuovo farmaco o una nuova indicazione terapeutica per una medicina già approvata viene esaminata da uno specifico organismo, chiamato Committee for Medicinal Products for Human Use (CHMP), composto dai rappresentanti dei 28 paesi dell'Unione Europea. La procedura di valutazione da parte del CHMP non deve durare più di 270 giorni. In caso di risposta negativa, l'industria farmaceutica ha il diritto di presentare un ricorso; mentre in caso di risposta positiva, tutti i paesi dell'UE sono obbligati a commercializzare il farmaco su richiesta dell'azienda.
L’EMA ha anche il compito di rimuovere i farmaci dal mercato quando gli effetti tossici sono maggiori dei benefici oppure quando i medicinali non presentano la stessa efficacia accertata al momento dell'autorizzazione. Purtroppo, gli effetti tossici dei farmaci si registrano sempre con ritardo, così succede spesso che la casa farmaceutica ritira il prodotto prima ancora che l'organismo regolatorio lo richiami. Esiste, poi, un’altra agenzia regolatoria che opera solo nostro Paese: l’AIFA - Agenzia Italiana del Farmaco. Le funzioni svolte da questo ente sono: autorizzare la commercializzazione di un farmaco, in Italia funzione del tutto amministrativa; inserire un medicinale nel prontuario terapeutico del Servizio Sanitario Nazionale, funzione puramente discrezionale, in quanto la decisione di pagare o meno per un determinato farmaco è lasciata ai singoli paesi. Unico neo nella gestione di queste pratiche da parte dell’EMA è l’impossibilità di accedere ai dati originali su cui basa le sue decisioni, a differenza di quanto succede negli Stati Uniti dove la FDA - Food and Drug Administration - mette a disposizione la documentazione relativa a tutto il processo di valutazione. Rendere disponibili i dati farmacologici, tossicologici e clinici è corretto nei confronti dei pazienti, della comunità scientifica e del Sistema Sanitario Nazionale.
Uso Compassionevole e Prescrizioni Off-Label
L’uso compassionevole non è riservato a malati in fase terminale, ma può essere utile in alcuni casi estremamente selezionati. Esiste un regolamento molto complesso a tutela dei malati nel caso in cui desiderino assumere, al di fuori di una sperimentazione clinica, un farmaco che non è ancora giunto alla fine del percorso di studio. Il medico curante deve inoltrare una richiesta dettagliata alla casa farmaceutica e chiedere una fornitura del farmaco. Sono inoltre necessari il nulla osta del comitato etico dell’ospedale e del paziente, che riceve spiegazioni sui pro e i contro della cura, ed è tenuto a firmare un consenso informato. Il farmaco deve essere stato inserito preventivamente in un elenco, presso l’Agenzia Italiana del Farmaco (AIFA), e deve avere superato la fase III di sperimentazione. Solo molto raramente si concede l’uso di una sostanza che si trova in una fase di sperimentazione più precoce.
Molti farmaci usati in oncologia non sono registrati per tutte le indicazioni possibili. Questo perché per ogni indicazione l’azienda dovrebbe effettuare appositi studi, molto costosi, che non sempre è conveniente portare avanti. In altri casi, la scienza progredisce più rapidamente della burocrazia, per cui vi sono sostanze di provata efficacia contro certi tumori che però non sono registrati per quella indicazione. Dare un farmaco fuori indicazione vuol dire fare la cosiddetta prescrizione off-label, che in inglese significa ‘fuori dall’etichetta’. Chiaramente non si devono prescrivere farmaci off-label, proprio per tutelare i malati dall’uso imprudente di alcune sostanze da parte dei medici. Ci sono però delle eccezioni, per esempio i tumori rari, per i quali difficilmente vengono registrati nuovi farmaci. Nel caso in cui si voglia prescrivere un farmaco off-label bisogna che l’opportunità di tale prescrizione venga valutata da commissioni ospedaliere indipendenti dal medico che la propone e, in certi casi, dal comitato di bioetica.
L'Innovazione nell'Industria Farmaceutica: L'Intelligenza Artificiale
L’intelligenza artificiale è la nuova frontiera della sperimentazione dei medicinali, che non richiederà più pazienti in carne e ossa. L'utilizzo dell'intelligenza artificiale (IA) può velocizzare la scoperta, lo sviluppo e le fasi di test dei nuovi medicinali, fino alla loro approvazione e commercializzazione, perché ha capacità uniche nell’integrazione di enormi quantità di dati provenienti dalla pratica clinica o da precedenti sperimentazioni. L'IA è una soluzione a diverse sfide che si hanno nella ricerca clinica, come l'accorciare i tempi in modo da rendere disponibili i farmaci in commercio il prima possibile e migliorare la produttività di ricerca e sviluppo. Oggi, solo il 10% dei medicinali che entrano negli studi clinici arrivano in commercio. La possibilità, attraverso l'interrogazione di database, la produzione di algoritmi e le simulazioni su modelli non umani, di aumentare il successo dell'intero processo di ricerca è cruciale, innanzitutto per i pazienti.
Ad esempio, sono stati sviluppati "digital twin", pazienti virtuali che contengono tutti i dati raccolti nei decenni su una specifica patologia e i suoi trattamenti. Nel caso dell'asma, un importante database di dati e informazioni consente di fare delle simulazioni nelle quali è possibile accelerare il passaggio dalla fase 1 alla fase 2 della sperimentazione, accorciando l’iter ordinario (laboratorio e primo gruppo ristretto di pazienti) e guadagnando un anno di tempo.
