Le cardiopatie congenite (CC) rappresentano un gruppo eterogeneo di patologie caratterizzate da alterazioni strutturali del cuore o dei grossi vasi, già presenti durante la vita fetale. Si chiamano congenite proprio perché sono malformazioni presenti fin dalla nascita, originate da un'anomalia nella formazione e nello sviluppo del cuore che si verifica durante la vita embrionale e fetale, in un periodo cruciale compreso tra la seconda e la nona settimana di gestazione. Queste malformazioni sono diverse per genere ed entità e, di conseguenza, impattano in modo differente sulla vita delle persone.
Le CC costituiscono la malformazione più comune alla nascita, interessando circa il 40% di tutti i difetti congeniti, e colpiscono approssimativamente un neonato ogni 100 nati vivi in Italia. La loro importanza clinica è notevole, poiché, tra i difetti di nascita, la cardiopatia congenita è la principale causa di mortalità infantile. Grazie ai recenti progressi della medicina, oggi è possibile fare una diagnosi accurata già durante la vita fetale, permettendo un approccio tempestivo e personalizzato alla cura di questi piccoli pazienti. Molti pazienti necessitano ogni anno di cure specialistiche cardiologiche e talvolta cardiochirurgiche, anche fino all'età adulta, mentre altri possono condurre una vita considerata "normale" a seconda della gravità e della gestione della loro condizione. I difetti strutturali possono coinvolgere diverse parti del cuore, come le pareti, le valvole, i vasi sanguigni o altre componenti essenziali per il corretto funzionamento dell'organo.
Cosa Sono le Cardiopatie Congenite?
Le cardiopatie congenite sono intrinsecamente malformazioni anatomiche del cuore, le cui origini sono da ricercarsi in uno sviluppo cardiaco incompleto o non corretto che si manifesta durante le prime settimane della vita embrionale. Queste anomalie sono presenti già nella vita intrauterina e la loro variazione in termini di tipo e gravità è estremamente ampia. La definizione stessa di cardiopatia congenita sottolinea la loro natura: esse sono alterazioni strutturali del cuore o dei grandi vasi che coesistono con l'individuo fin dal momento della sua nascita. Questo gruppo di patologie è estremamente diversificato, comprendendo difetti che possono essere molto semplici e avere un impatto minimo sulla salute del neonato, fino a malformazioni complesse che richiedono interventi medici immediati e sofisticati.
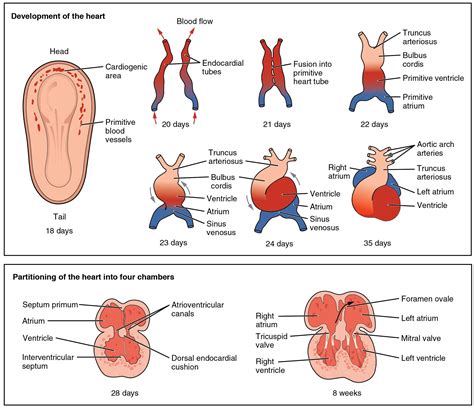
I difetti possono interessare le pareti che dividono le camere cardiache, le valvole che regolano il flusso sanguigno, i grandi vasi che entrano ed escono dal cuore, o una combinazione di queste componenti. La precoce insorgenza di queste anomalie, tra la seconda e la nona settimana di gestazione, evidenzia la delicatezza del processo di formazione del cuore e la potenziale vulnerabilità a fattori che possono interferire con il suo corretto sviluppo. Comprendere la genesi e la natura di queste malformazioni è il primo passo fondamentale per la loro diagnosi e un'efficace gestione terapeutica.
Epidemiologia e Prevalenza in Italia
Le cardiopatie congenite rappresentano, senza dubbio, la malformazione più comune rilevata alla nascita, con una prevalenza che si attesta intorno al 40% tra tutti i difetti congeniti. In Italia, dati recenti indicano che circa un neonato ogni 100 nati vivi è affetto da una qualche forma di cardiopatia congenita. Questa frequenza le rende una delle principali preoccupazioni in ambito pediatrico e neonatale.
È significativo notare che circa l'80% delle cardiopatie congenite vengono scoperte nel primo anno di vita, con una percentuale ancora più elevata, circa il 50%, che emerge già nel primo mese di vita del neonato. Questa tempestività nella diagnosi è cruciale per la gestione di molte condizioni. Le cardiopatie congenite sono le anomalie congenite più comuni, verificandosi in quasi l'1% dei nati vivi (1). Tra i difetti di nascita, la cardiopatia congenita è la principale causa di mortalità infantile, sottolineando l'urgenza e l'importanza di una diagnosi e un trattamento precoci.
Le più comuni malattie cardiache congenite diagnosticate nella prima infanzia includono i difetti del setto interventricolare, sia di tipo muscolare che perimembranoso, seguiti dai difetti del setto interatriale di tipo ostium secundum. La prevalenza totale di queste condizioni è di 48,4 su 10.000 nati vivi (2, 3, 4). Per quanto riguarda le cardiopatie cianogene, la più comune è la tetralogia di Fallot, la quale si manifesta con una prevalenza doppia rispetto alla trasposizione dei grossi vasi (4,7 contro 2,3 ogni 10.000 nascite). In un quadro più ampio, le valvole aortiche bicuspidi costituiscono i difetti congeniti più frequenti in assoluto, con una prevalenza riportata che varia dallo 0,5% al 2,0% della popolazione generale. Questi dati epidemiologici evidenziano l'ampio spettro e la significativa incidenza di queste patologie, rendendo la loro comprensione e gestione una priorità sanitaria.
Le Cause delle Cardiopatie Congenite: Un'Eziologia Multifattoriale
L'origine delle cardiopatie congenite è complessa e viene definita come multifattoriale, riconoscendo cioè l'interazione di diversi elementi. La ricerca ha evidenziato che complessi fattori ambientali e genetici contribuiscono allo sviluppo di queste patologie. Sebbene in una piccola percentuale dei casi siano implicate cause ben definite, la maggior parte delle volte si tratta di una combinazione di predisposizioni.

Tra i fattori ambientali comuni che possono influenzare lo sviluppo cardiaco fetale, si annoverano diverse condizioni materne e specifiche esposizioni. Le patologie materne rivestono un ruolo significativo; ad esempio, il diabete mellito gestazionale o preesistente, l'obesità materna, e alcune infezioni virali come la rosolia e l'influenza, sono stati identificati come fattori di rischio. Anche il lupus eritematoso sistemico materno può contribuire a tali anomalie. Deficit nutrizionali, come una carenza di acido folico o di vitamine A e D, durante la gravidanza sono ugualmente considerati fattori predisponenti. Inoltre, l'esposizione a sostanze teratogene è un rischio accertato; ciò include l'assunzione di alcool, il fumo di sigaretta, e l'uso di specifici farmaci come la talidomide, il litio e alcuni anticonvulsivanti, così come l'esposizione a sostanze chimiche o a radiazioni. L'età materna è un noto fattore di rischio per alcune condizioni genetiche specifiche, in particolare la Sindrome di Down, la quale spesso include difetti cardiaci. Tuttavia, non è ancora del tutto chiaro se l'età materna rappresenti un fattore di rischio indipendente direttamente per la cardiopatia congenita, al di fuori del contesto delle sindromi genetiche associate. Anche l'età paterna è stata oggetto di studi e può essere considerata un potenziale fattore di rischio (1).
Accanto ai fattori ambientali, la ricerca ha progressivamente sottolineato come nella patogenesi multifattoriale delle cardiopatie congenite giochino sempre più un ruolo accertato le cause genetiche, attraverso meccanismi complessi ed eterogenei.Alcune anomalie cromosomiche numeriche, note come aneuploidie, sono fortemente associate a cardiopatie congenite. Tra queste, la trisomia 21 (meglio conosciuta come sindrome di Down), la trisomia 18, la trisomia 13 e la monosomia X (sindrome di Turner) sono le più rilevanti. È importante sottolineare, tuttavia, che queste anomalie genetiche rappresentano solo una quota del 5-6% dei pazienti con cardiopatie congenite.
La maggior parte degli altri casi con una componente genetica coinvolge delezioni subcromosomiche (microdelezioni), duplicazioni subcromosomiche o mutazioni di un singolo gene. Spesso, queste mutazioni sono responsabili di sindromi congenite che interessano non solo il cuore, ma anche altri organi. Alcuni esempi ben noti includono la sindrome di DiGeorge, causata da una microdelezione nella regione 22q11.2 del cromosoma, e la sindrome di Williams (talvolta denominata sindrome di Williams-Beuren), associata a una microdelezione nella regione 7p11.23. Difetti di un singolo gene possono anche causare sindromi specifiche associate a cardiopatie congenite, come le mutazioni di fibrillina-1 nella sindrome di Marfan, TXB5 nella sindrome di Holt-Oram e PTPN11 nella sindrome di Noonan. È anche possibile che difetti di un singolo gene siano alla base di difetti cardiaci congeniti isolati, ovvero non sindromici.
Nonostante l'avanzamento delle conoscenze genetiche, in circa il 72% dei pazienti con cardiopatia congenita non viene rilevata alcuna eziologia genetica identificabile (2, 3, 4), evidenziando la complessità della patologia e la necessità di ulteriori ricerche.
L'importanza clinica di una diagnosi precoce, soprattutto quando supportata dalla conferma genetica di un quadro sindromico, è che essa può guidare lo specialista alla ricerca di una particolare cardiopatia congenita. Questo permette di anticiparne la storia naturale, valutarne l'outcome prognostico (che può differire tra pazienti sindromici e non a parità di cardiopatia), e di stabilire protocolli di follow-up multidisciplinari personalizzati. Il rischio di recidiva delle cardiopatie congenite all'interno di una famiglia varia in funzione della causa sottostante. Questo rischio è trascurabile nelle mutazioni de novo, si attesta tra il 2% e il 5% nelle cardiopatie congenite multifattoriali non sindromiche e può raggiungere il 50% quando la causa è una mutazione autosomica dominante. L'identificazione di una valvola aortica bicuspide in un individuo merita uno screening familiare, data la prevalenza familiare riportata del 9% (5). È fondamentale identificare i fattori genetici, specialmente perché un numero sempre maggiore di pazienti con cardiopatie congenite, rispetto al passato, riesce a sopravvivere fino all'età adulta e, potenzialmente, a formare una propria famiglia.
Prevenzione: Misure per Ridurre il Rischio
La prevenzione delle cardiopatie congenite, sebbene non sempre possibile data la complessità multifattoriale delle cause, si concentra principalmente sulla riduzione dei fattori di rischio ambientali e materni modificabili durante la gravidanza. La Società Italiana di Neonatologia (SIN) e la Società Italiana di Cardiologia Pediatrica e delle Cardiopatie Congenite (SICP) hanno fornito importanti informazioni utili volte ad adottare un corretto percorso di prevenzione, diagnosi e cura di tali patologie, soprattutto in occasione di eventi come la Giornata Mondiale delle cardiopatie congenite.
Neonatologi ed esperti in cardiologia pediatrica hanno evidenziato che l'adozione di stili di vita appropriati durante l'intera gravidanza è un fattore chiave per prevenire e ridurre il rischio di comparsa di malformazioni congenite. Questo include, in particolare, l'assoluto divieto di consumo di alcool e fumo, così come l'evitamento di alcuni farmaci che sono noti per avere effetti teratogeni.
Un altro aspetto fondamentale della prevenzione nutrizionale è una dieta ricca di acido folico. L'integrazione di acido folico, soprattutto nel periodo preconcezionale e nelle prime fasi della gravidanza, è universalmente raccomandata per la prevenzione di numerosi difetti del tubo neurale e, indirettamente, può avere un ruolo nella riduzione del rischio di alcune cardiopatie.
Infine, la vaccinazione contro le principali malattie infettive a rischio teratogeno, come ad esempio la rosolia, è un'efficace misura preventiva. L'immunizzazione materna pre-gravidanza è cruciale per proteggere il feto da infezioni che possono gravemente compromettere il suo sviluppo, inclusa la formazione cardiaca. Adottare queste strategie preventive può contribuire significativamente a un esito di gravidanza più sano e a una riduzione dell'incidenza delle cardiopatie congenite.
Diagnosi delle Cardiopatie Congenite: Dal Feto al Neonato
La diagnosi delle cardiopatie congenite ha fatto passi da gigante negli ultimi anni, permettendo un'identificazione sempre più precoce e precisa, sia in epoca prenatale che postnatale. Questa tempestività è fondamentale per ottimizzare le strategie di cura.
Diagnosi Prenatale
L'ecocardiografia fetale è un esame strumentale che rende attualmente possibile, con elevata attendibilità, la diagnosi prenatale delle principali malformazioni cardiache sul feto. Con i recenti progressi della medicina, è infatti possibile fare diagnosi durante la vita fetale con un semplice ecocardiogramma fetale. Questo esame, che richiede circa 30 minuti di tempo, o più se la cardiopatia è complessa, è in grado di assicurare una diagnosi completa e dettagliata di qualsiasi cardiopatia congenita.
L'importanza clinica della diagnosi prenatale è immensa: se l'ecografia prenatale rileva il sospetto di una cardiopatia congenita critica, diviene opportuno pianificare il parto in Centri specializzati. Questi centri devono essere forniti di reparti di cardiologia pediatrica, terapia intensiva cardiologica e cardiochirurgia pediatrica. Tale pianificazione anticipata consente di affrontare al meglio l'evento nascita e le prime ore di vita del neonato affetto. In alcune circostanze, la diagnosi prenatale può addirittura portare alla proposta di induzione del parto o di un parto con taglio cesareo, sempre nell'ottica di garantire le migliori condizioni assistenziali.
Ecocardiografia Fetale 2 - Corso Avanzato Esclusiva
Diagnosi Postnatale
Nei casi in cui la diagnosi non è stata stabilita in epoca prenatale, è possibile diagnosticare le cardiopatie congenite nelle prime 48-72 ore di vita del neonato attraverso una precisa anamnesi ostetrica, che indaga su eventuali infezioni, diabete o assunzione di farmaci da parte della madre, e un accurato esame clinico del neonato. Circa l'80% delle cardiopatie congenite si scoprono nel primo anno di vita e il 50% circa nel primo mese di vita, sottolineando l'importanza di un'attenta osservazione post-nascita. Alcune cardiopatie devono essere diagnosticate, curate e operate immediatamente in epoca neonatale, evidenziando l'urgenza di un riconoscimento rapido.
I sintomi che possono ricondurre alla diagnosi di cardiopatie congenite sono vari e dipendono dalla specifica malformazione e dalla sua gravità. È importante sottolineare che la presenza e la gravità dei sintomi dipendono dalla specifica condizione cardiaca congenita e dalla sua gestione. Tra i segnali più comuni riscontrati nel neonato vi sono il pallore, la cianosi (colorazione bluastra della pelle e delle mucose), un respiro veloce e difficoltoso (dispnea e tachipnea), un ritmo cardiaco accelerato (tachicardia) e la presenza di soffi cardiaci. Questo soffio, che è un suono anomalo rilevato all'auscultazione del cuore, viene riscontrato frequentemente nei primi giorni o nelle prime settimane di vita. Nel sospetto o dopo diagnosi di cardiopatia congenita, è opportuno rivolgersi a un cardiologo pediatra o, ancora meglio, a un Centro Specializzato, dove il piccolo paziente possa essere controllato per tutto il percorso diagnostico e di cura farmacologica e chirurgica.
L'ecocardiografia, sia transtoracica che in alcuni casi transesofagea, è lo strumento diagnostico fondamentale. L'ecocardiografia consente la diagnosi finale di tale patologia, in quanto capace di identificare dettagliatamente il tipo di anomalia presente e di definire le opzioni di trattamento. Le indagini strumentali a disposizione sono in grado di assicurare una diagnosi completa e dettagliata di qualsiasi cardiopatia congenita. Nel caso dell'ecocardiografia transesofagea, una sonda ecografica viene introdotta nell'esofago. Poiché l'esofago è molto vicino al cuore, si possono ottenere immagini molto dettagliate di svariate strutture del cuore, compresa quella delle valvole cardiache. Per queste due indagini invasive (se si considera invasiva anche la transesofagea per le implicazioni) è necessaria l'autorizzazione dei genitori (consenso informato). Sebbene la maggioranza dei casi sia diagnosticata nei primi mesi di vita, talvolta alcune cardiopatie possono rimanere asintomatiche fino all'età adulta, rendendo la diagnosi un processo che può estendersi nel tempo.
La Circolazione Fetale e la Transizione alla Nascita
Per comprendere appieno le cardiopatie congenite, è fondamentale avere chiarezza sul funzionamento della circolazione fetale e sulle profonde trasformazioni che avvengono alla nascita per stabilire la circolazione adulta.
Circolazione Fetale Normale
La circolazione nel feto è intrinsecamente diversa da quella postnatale, essendo caratterizzata dalla presenza di shunt che bypassano i polmoni, non ancora attivi nella funzione respiratoria. Il sangue che entra nel lato destro del cuore del feto è già stato ossigenato attraverso la placenta. Poiché i polmoni non sono ventilati e non sono quindi in grado di scambiare ossigeno e anidride carbonica, solo una piccola quantità di sangue deve passare attraverso l'arteria polmonare. La maggior parte del sangue dal lato destro del cuore bypassa i polmoni attraverso due strutture fondamentali: il forame ovale e il dotto arterioso.
La circolazione fetale è caratterizzata da uno shunt destro-sinistro di sangue attorno ai polmoni non ventilati. Questo shunt avviene principalmente attraverso il dotto arterioso pervio, che connette l'arteria polmonare all'aorta, e il forame ovale, che mette in comunicazione l'atrio destro e quello sinistro. Tale shunt è favorito dalle alte resistenze arteriolari polmonari, causate dalla non espansione dei polmoni e dall'ipossia relativa, e dalla resistenza al flusso ematico relativamente bassa nel circolo sistemico, inclusa la placenta. Di conseguenza, circa il 90-95% del flusso in uscita dal cuore destro bypassa i polmoni e raggiunge direttamente la circolazione sistemica.
Il dotto arterioso fetale è mantenuto aperto da una bassa PaO2 sistemica fetale, che si aggira intorno ai 25 mmHg, insieme alle prostaglandine prodotte localmente. Il forame ovale, a sua volta, è mantenuto aperto dai gradienti delle pressioni atriali: nel feto, la pressione atriale sinistra è relativamente bassa a causa del ridotto ritorno ematico polmonare, mentre la pressione atriale destra è relativamente alta a causa del notevole ritorno ematico dalla placenta.
Le frecce rosse in uno schema rappresentano il sangue fetale più ossigenato, con una saturazione di ossigeno uguale o superiore al 65%. Le frecce blu indicano il sangue meno ossigenato, con una saturazione di ossigeno uguale o inferiore al 45%. Le frecce viola denotano una saturazione di ossigeno intermedia, tipicamente tra il 50% e il 60%. È importante notare che la saturazione di ossigeno nel feto è significativamente inferiore rispetto ai valori che si riscontrano durante la vita postnatale.
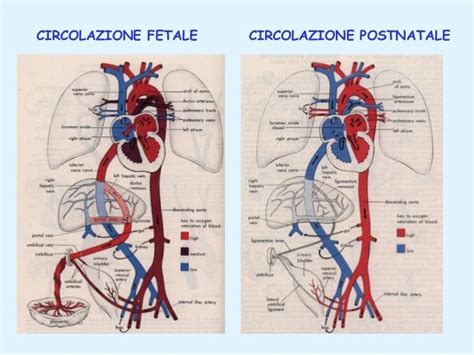
Cambiamenti Perinatali
Con i primi atti respiratori del neonato, si verificano profonde e rapide modificazioni a livello della circolazione sistemica e polmonare, che portano alla chiusura delle strutture fetali e all'instaurazione del modello circolatorio adulto.
Il primo e più significativo cambiamento è un aumento massivo del flusso sanguigno polmonare. Questo è reso possibile da una caduta improvvisa delle resistenze arteriolari polmonari. Tale riduzione è il risultato diretto della vasodilatazione che deriva dall'espansione polmonare (i polmoni si riempiono d'aria), dall'aumentata PaO2 (l'ossigeno viene ora assorbito dai polmoni) e dalla ridotta PaCO2. Le forze elastiche delle coste e della parete toracica diminuiscono la pressione interstiziale polmonare, determinando un ulteriore aumento del flusso ematico attraverso i capillari polmonari.
Contemporaneamente, si verifica la chiusura funzionale del forame ovale. L'aumentato ritorno venoso dai polmoni incrementa la pressione atriale sinistra, mentre la pressione atriale destra diminuisce a causa della cessazione del flusso ematico placentare. Il ridotto flusso ematico placentare si traduce in una riduzione o cessazione del ritorno ematico all'atrio destro. Così, la pressione atriale destra diminuisce mentre aumenta la pressione atriale sinistra; di conseguenza, i due componenti fetali del setto interatriale (septum primum e septum secundum) vengono accostati, arrestando il flusso attraverso il forame ovale. Questo differenziale di pressione riduce la pressione differenziale tra atrio sinistro e destro, contribuendo alla chiusura funzionale del forame ovale. Nella maggior parte delle persone, i due setti alla fine si fondono e il forame ovale cessa di esistere. Tuttavia, nel 25% degli adulti, il forame ovale può rimanere pervio con uno shunt residuo minimo o nullo (1).
Subito dopo la nascita, le resistenze sistemiche superano quelle polmonari. Si verifica, quindi, un'inversione rispetto alla situazione fetale e la direzione del flusso ematico attraverso il dotto arterioso pervio si inverte, creando uno shunt ematico sinistro-destro. Questa situazione, definita circolazione di transizione, persiste da subito dopo la nascita fino a circa 24-72 ore di vita, momento in cui il dotto arterioso inizia a comprimersi e a chiudersi. Il sangue proveniente dall'aorta che entra nel dotto arterioso e nei suoi vasa vasorum ha un'alta PO2 che, insieme alle variazioni nel metabolismo delle prostaglandine, determina la vasocostrizione e la chiusura del dotto arterioso. Una volta che il dotto arterioso si chiude definitivamente, si instaura la circolazione di tipo adulto. A questo punto, i due ventricoli pompano in serie e non esistono più shunt rilevanti tra la circolazione polmonare e quella sistemica.
Ritorno alla Circolazione Fetale (Distress)
Durante i primi giorni di vita, in caso di distress neonatale grave, si può assistere a un preoccupante ritorno a una circolazione di tipo fetale. Situazioni di asfissia con ipossia e ipercapnia causano una vasocostrizione delle arteriole polmonari, elevando nuovamente le resistenze polmonari. Contemporaneamente, si verifica una vasodilatazione del dotto arterioso, con un'inversione dei processi descritti precedentemente. Ciò porta alla realizzazione di uno shunt destro-sinistro attraverso il dotto arterioso, che diventa nuovamente pervio, e/o il forame ovale, che si riapre. Di conseguenza, il neonato presenta una grave ipossiemia, una condizione che viene denominata ipertensione polmonare persistente o circolazione fetale persistente, nonostante non vi sia più la circolazione ombelicale. In questi casi, l'obiettivo del trattamento è invertire le condizioni che hanno causato la vasocostrizione polmonare, ripristinando la normale circolazione postnatale.
Fisiopatologia delle Anomalie Cardiache Congenite
Le anomalie cardiache congenite sono classificate in base alle loro implicazioni emodinamiche e alla presenza di cianosi. Questa classificazione aiuta a comprendere le diverse manifestazioni cliniche e i meccanismi sottostanti. Le principali categorie sono: cardiopatie cianogene e cardiopatie non cianogene, che includono shunt sinistro-destro o lesioni ostruttive. Le conseguenze fisiologiche di queste anomalie variano notevolmente, potendo spaziare da un semplice soffio cardiaco o una discrepanza dei polsi in un bambino asintomatico, fino a condizioni estreme come cianosi grave, insufficienza cardiaca o collasso circolatorio, evidenziando l'ampio spettro di presentazione clinica.
Cardiopatie Cianogene
Nelle cardiopatie cianogene, quantità variabili di sangue venoso deossigenato sono deviate al cuore sinistro attraverso uno shunt destro-sinistro. Questo meccanismo riduce la saturazione arteriosa sistemica di ossigeno, portando alla caratteristica colorazione bluastra della pelle e delle mucose, nota come cianosi. La cianosi diventa clinicamente evidente quando la concentrazione di emoglobina deossigenata supera i 5 g/dL (o 50 g/L).
Una cianosi persistente e non trattata può portare a diverse complicanze significative. Tra queste, si annoverano la policitemia (un aumento anomalo dei globuli rossi, nel tentativo di compensare la carenza di ossigeno), l'ippocratismo digitale (dita a bacchetta di tamburo), il tromboembolismo (inclusi ictus cerebrali, dovuti alla maggiore viscosità del sangue e alla possibilità di passaggio di emboli dal lato destro al sinistro del cuore), disturbi emorragici, ascesso cerebrale e iperuricemia. Nei neonati affetti da tetralogia di Fallot non riparata o da altri difetti congeniti complessi che presentano una stenosi sottopolmonare dinamica e un difetto del setto ventricolare, possono verificarsi crisi ipercianotiche, episodi acuti di grave desaturazione e peggioramento della cianosi.
A seconda della specifica malformazione, il flusso ematico polmonare nelle cardiopatie cianogene può essere ridotto, normale o aumentato, spesso sfociando in insufficienza cardiaca in aggiunta alla cianosi stessa, e la cianosi può essere di variabile entità. I soffi cardiaci, sebbene variabilmente auscultabili, non sono specifici di queste condizioni e la loro presenza o assenza non è sufficiente per una diagnosi precisa.
Shunt Sinistro-Destro
Gli shunt sinistro-destro rappresentano un'altra categoria significativa di cardiopatie congenite non cianogene. In queste condizioni, il sangue ossigenato proveniente dal cuore sinistro (atrio sinistro o ventricolo sinistro) o dall'aorta viene deviato al cuore destro (atrio destro o ventricolo destro) o all'arteria polmonare attraverso una breccia o una comunicazione anomala tra i due lati della circolazione.
Immediatamente dopo la nascita, la resistenza vascolare polmonare è ancora relativamente elevata, e di conseguenza, il flusso attraverso questa comunicazione può essere minimo o bidirezionale. Tuttavia, entro le prime 24-48 ore di vita, la resistenza vascolare polmonare scende progressivamente. A questo punto, il sangue inizierà sempre più a scorrere da sinistra a destra, dato che la pressione nel cuore sinistro e nell'aorta è maggiore rispetto a quella del cuore destro e dell'arteria polmonare.
L'ulteriore afflusso di sangue al lato destro del cuore aumenta il flusso ematico polmonare e la pressione arteriosa polmonare in vario grado. Maggiore è l'aumento di questo flusso e pressione, più gravi saranno i sintomi e i segni clinici. Al contrario, un piccolo shunt sinistro-destro tipicamente non provoca sintomi o segni significativi.
Gli shunt ad alta pressione, come quelli che si verificano a livello ventricolare (ad esempio, ampi difetti del setto interventricolare) o delle grandi arterie (come un dotto arterioso pervio), tendono a manifestarsi clinicamente diversi giorni o alcune settimane dopo la nascita. Al contrario, gli shunt a bassa pressione, quali i difetti del setto interatriale, compaiono considerevolmente più tardi, spesso nell'infanzia o anche in età adulta, a causa delle minori differenze di pressione tra gli atri.
Se non trattati, l'elevato flusso ematico polmonare e la pressione arteriosa polmonare che ne derivano possono portare a malattia vascolare polmonare, una condizione irreversibile che danneggia i vasi sanguigni dei polmoni, e infine alla sindrome di Eisenmenger, uno stadio avanzato in cui lo shunt può invertirsi, causando cianosi. Grandi shunt sinistro-destro, come un ampio difetto del setto interventricolare o un dotto arterioso pervio, causano un eccesso di flusso sanguigno polmonare e un sovraccarico di volume del ventricolo sinistro. Questo può portare a segni di insufficienza cardiaca che, durante la prima infanzia, spesso si traducono in difficoltà di crescita. Inoltre, un ampio shunt sinistro-destro determina una minore compliance polmonare e una maggiore resistenza delle vie respiratorie. Questi fattori aumentano la probabilità di ospedalizzazione nei neonati con infezioni da virus respiratorio sinciziale (RSV) o altre infezioni del tratto respiratorio superiore o inferiore, rendendoli particolarmente vulnerabili.
Lesioni Ostruttive
Le lesioni ostruttive rappresentano una categoria di cardiopatie congenite in cui il flusso sanguigno all'interno o all'esterno del cuore è impedito o ristretto. Questa ostruzione crea un gradiente pressorio significativo attraverso il punto stenotico. Il risultato di tale ostruzione è un sovraccarico pressorio a monte della restrizione, il quale può condurre a ipertrofia del ventricolo interessato, che lavora di più per superare l'ostacolo, e, se non trattato, può evolvere in insufficienza cardiaca.
La manifestazione più ovvia e spesso rilevata per la prima volta in queste condizioni è un soffio cardiaco. Questo suono anomalo è generato dal flusso turbolento del sangue che attraversa il punto ristretto o ostruito (stenotico). Esempi comuni di lesioni ostruttive includono la stenosi aortica congenita, che costituisce circa il 3-6% delle cardiopatie congenite, e la stenosi polmonare congenita, che rappresenta una percentuale maggiore, tra l'8% e il 12% dei casi (1, 2). La gravità dell'ostruzione determina l'entità del gradiente di pressione e, di conseguenza, la severità dei sintomi e la necessità di un intervento.
Insufficienza Cardiaca
L'insufficienza cardiaca è una complicanza grave che può derivare da alcune cardiopatie congenite. È importante notare che non tutte le cardiopatie causano un'alterazione significativa dell'emodinamica; ad esempio, una valvola aortica bicuspide o una stenosi aortica lieve possono non influenzare in modo sostanziale la funzione cardiaca per lungo tempo. Tuttavia, altre anomalie cardiache congenite sono tali da causare un sovraccarico di pressione o di volume, elementi che, se prolungati, possono culminare in insufficienza cardiaca.
L'insufficienza cardiaca si verifica quando la gittata cardiaca, ovvero la quantità di sangue che il cuore pompa nell'arco di un minuto, è insufficiente a soddisfare le esigenze metaboliche dell'organismo. Alternativamente, può manifestarsi quando il cuore non riesce a gestire adeguatamente il ritorno venoso. Questa incapacità del cuore di funzionare in modo efficiente porta a diverse conseguenze cliniche. Nel caso di insufficienza del ventricolo sinistro, si può avere congestione polmonare, con accumulo di liquidi nei polmoni, causando difficoltà respiratorie. Nell'insufficienza del ventricolo destro, si manifesta un accumulo di liquidi soprattutto nei tessuti dipendenti, come gli arti inferiori, e nei visceri addominali, portando a edema e gonfiore. Spesso, l'insufficienza cardiaca può interessare entrambi i ventricoli, con un quadro clinico combinato. Nel neonato, i segni di insufficienza cardiaca possono essere sottili e includere difficoltà nell'alimentazione, scarsa crescita ponderale, tachipnea e sudorazione eccessiva.
Trattamento e Gestione delle Cardiopatie Congenite
Il trattamento delle cardiopatie congenite è un percorso altamente specializzato e dipende in modo critico dal tipo e dalla gravità della specifica condizione diagnosticata. Non esiste un approccio unico, ma piuttosto un piano terapeutico personalizzato, studiato attentamente in base alle esigenze individuali del paziente.
Alcuni difetti cardiaci congeniti possono essere di entità tale da richiedere solo un monitoraggio regolare e attento. In questi casi, il team medico seguirà l'evoluzione della condizione per assicurarsi che non si verifichino peggioramenti o complicanze che richiedano un intervento più aggressivo. Questo tipo di gestione è spesso applicato a difetti minori che possono risolversi spontaneamente o che non compromettono significativamente la funzione cardiaca.
Per altre cardiopatie, la gravità della malformazione o le sue implicazioni emodinamiche rendono necessari interventi più invasivi. Questi possono includere interventi chirurgici correttivi o palliativi, procedure invasive come il cateterismo cardiaco (che può essere sia diagnostico che terapeutico, permettendo di riparare alcuni difetti senza la necessità di un'operazione a cuore aperto), e in casi estremamente complessi e severi, anche il trapianto di cuore. Per molte di queste procedure, l'obiettivo è ripristinare il più possibile una normale funzione cardiaca o, in alternativa, ottimizzare la circolazione per garantire la migliore qualità di vita possibile.
Un aspetto cruciale nella gestione è la tempistica dell'intervento. Alcune cardiopatie congenite devono essere diagnosticate, curate e operate immediatamente in epoca neonatale, poiché rappresentano un pericolo imminente per la vita del bambino. Per altre condizioni, tuttavia, dopo la diagnosi neonatale, può essere utile attendere qualche settimana o addirittura qualche mese prima di procedere con l'intervento chirurgico. Questa attesa permette al neonato di crescere, di acquisire una maggiore stabilità fisiologica e può consentire di ottenere i migliori risultati chirurgici, riducendo i rischi associati all'operazione in un organismo estremamente piccolo e fragile.
Nel sospetto o dopo la diagnosi di cardiopatia congenita, è di fondamentale importanza rivolgersi a un cardiologo pediatra. Meglio ancora, il piccolo paziente dovrebbe essere indirizzato a un Centro Specializzato ove possa essere controllato e seguito per tutto il percorso diagnostico e di cura, che può includere terapie farmacologiche specifiche oltre agli interventi chirurgici. Questi centri offrono un approccio multidisciplinare, con la collaborazione di cardiologi pediatrici, cardiochirurghi pediatrici, neonatologi e altri specialisti, garantendo una gestione completa e integrata delle complesse esigenze di questi bambini. Le Società Italiana di Neonatologia (SIN) e di Cardiologia Pediatrica e delle Cardiopatie Congenite (SICP) evidenziano costantemente l'importanza di questi percorsi specialistici per garantire ai pazienti le migliori possibilità di cura e un futuro il più sano possibile.
tags: #cardiopatia #congenita #neonato