La gravidanza è un periodo di profonde trasformazioni fisiologiche nel corpo femminile, e tra queste, i cambiamenti a carico del sistema ematico rivestono un'importanza cruciale. L'assetto emoglobinico, ovvero l'analisi dei diversi tipi e delle quantità relative di emoglobina presenti nel sangue, rappresenta uno strumento diagnostico fondamentale per monitorare la salute della madre e del feto. Questo esame è un ausilio alla ricerca delle varianti emoglobiniche o di una classe di patologie note con il nome di talassemie, caratterizzate dalla diminuita produzione o carenza di una delle catene globiniche. La corretta interpretazione dei suoi valori, in particolare quelli dell'emoglobina A2 (Hb A2) e dell'emoglobina fetale (HbF), è essenziale per identificare precocemente eventuali condizioni di rischio, come anemie o emoglobinopatie, che potrebbero influenzare l'esito della gestazione e la salute del nascituro.
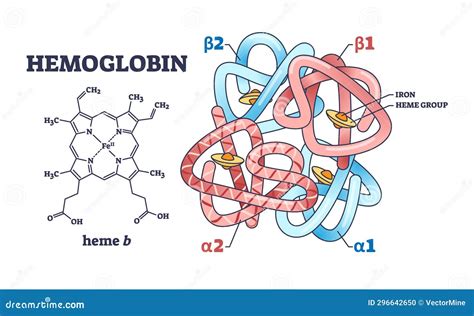
L'Emoglobina: Un Trasportatore Vitale e le Sue Varianti Fisiologiche
L'emoglobina (Hb) è una proteina contenuta all'interno dei globuli rossi, e il suo ruolo consiste nel trasportare l'ossigeno ai tessuti del nostro organismo. Questa proteina è formata da quattro subunità, chiamate globine o catene globiniche, ciascuna delle quali presenta al suo interno una struttura denominata gruppo eme, che contiene ferro ed è responsabile del legame dell'ossigeno. Esistono quattro tipi diversi di catene globiniche, designate come: α (alfa), β (beta), γ (gamma), δ (delta).
Le principali varianti fisiologiche dell'emoglobina nell'adulto sono l'Hb-A1, che rappresenta il 97-98% dell'emoglobina totale; l’emoglobina A2 (Hb-A2), che costituisce il 2-3% del totale, e l'HbF, o emoglobina fetale, che è l'emoglobina predominante nella vita fetale. Nell’emoglobina fetale, nello specifico, si trovano due catene α e due catene γ (alfa2 gamma2), mentre le emoglobine di un adulto sono composte da due coppie di catene α e ß (alfa2 beta2) per l'HbA1 e α2 δ2 per l'HbA2. Queste modificazioni strutturali conferiscono all'emoglobina fetale un'affinità per l'ossigeno superiore; in altre parole, si lega all'ossigeno in modo più tenace rispetto all'emoglobina adulta. Dal punto di vista funzionale, l'emoglobina fetale (HbF od emoglobina F) permette al feto di estrarre con maggiore efficacia l'ossigeno dal sangue materno. Riesce infatti a trasportare percentuali comprese tra il 20% e il 30% di ossigeno in più, rispetto all’emoglobina materna. Il trasferimento di ossigeno al sangue fetale attraverso la barriera placentare è favorito anche dalla maggiore concentrazione di emoglobina, più alta di circa il 50% rispetto a quella del sangue materno. La sintesi delle globine Beta caratterizzanti l'emoglobina adulta, appena percettibile durante la vita fetale, raggiunge il normale regime soltanto verso la fine del terzo mese della vita extrauterina.
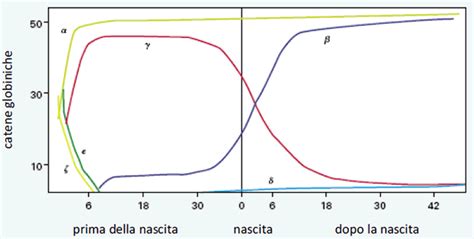
L'Emoglobina Fetale (HbF): Dalla Vita Fetale all'Età Adulta
L'emoglobina fetale (HbF) è la prima emoglobina prodotta durante la vita fetale. Nello specifico, l'emoglobina fetale è formata da due catene α e da due catene γ, costituite rispettivamente da 141 e 146 amminoacidi. Le due catene alfa sono identiche a quelle presenti nell'emoglobina adulta, mentre quelle gamma differiscono dalle Beta per 39 amminoacidi. Questa modifica strutturale conferisce all'emoglobina fetale un'affinità per l'ossigeno superiore, permettendo al feto di estrarre con maggiore efficacia l'ossigeno dal sangue materno.
Normalmente, la produzione di HbF diminuisce progressivamente alla nascita per poi stabilizzarsi su valori molto bassi intorno a 1-2 anni. Entro il primo anno di vita, le concentrazioni di emoglobina fetale scendono a livelli generalmente inferiori all'1%. Negli adulti normali, i valori di emoglobina fetale sono compresi tra lo 0.3% e l'1.2%.
L'esame per l’emoglobina fetale viene svolto per la diagnosi di patologie a carico del sangue, quali talassemia e anemia falciforme. Per l'esame dell'emoglobina fetale viene richiesto un digiuno di almeno otto ore. Tuttavia, a differenza di altri esami del sangue, come quelli per la glicemia o il colesterolo, per l’esame dell’HbF non è sempre richiesto il digiuno. È importante informare il medico di tutti i farmaci, integratori o terapie ormonali che si stanno assumendo, poiché alcuni di essi potrebbero influenzare i risultati dell’esame.
Quando negli adulti si hanno valori di emoglobina fetale superiori a 1.1, si parla di emoglobina F alta. Questa condizione può essere, ad esempio, una condizione di recupero dovuta a ipoplasia di midollo osseo, ovvero un disturbo delle cellule staminali ematopoietiche. Alcuni soggetti sono affetti dalla cosiddetta persistenza ereditaria dell'emoglobina fetale, una condizione benigna in cui concentrazioni importanti di emoglobina fetale (> 10%) persistono anche in età adulta. Si è notato come tale peculiarità, generalmente asintomatica, possa alleviare la severità di certe emoglobinopatie e talassemie. Una terapia farmacologica capace di aumentare la concentrazione di emoglobina fetale apporta benefici significativi ad alcune categorie di pazienti, come quelli affetti da anemia falciforme o da talassemia Beta. Il prototipo di questi farmaci è stata l'idrossiurea, farmaco antineoplastico ad azione mielosopressiva, che si è dimostrato efficace nell'aumentare i livelli di emoglobina fetale e nel ridurre l'incidenza di crisi dolorose in pazienti affetti da anemia falciforme.
ANEMIA IN GRAVIDANZA: i miei consigli
L'Emoglobina A2 (HbA2): Un Indicatore Cruciale nella Diagnostica
L'emoglobina A2 (Hb A2), costituita da due catene alfa e due catene delta (α2 δ2), è una frazione minore dell'emoglobina totale nell'adulto. Le raccomandazioni per la diagnostica di primo livello delle emoglobinopatie, anche nella recente revisione di prossima pubblicazione sul sito della Società Italiana Talassemie ed Emoglobinopatie (SITE), indicano l’intervallo di normalità dell’Hb A2 compreso tra 2,5 e 3,2%.
Il precedente "caso 10" di questo portale poneva in evidenza l’aumento abnorme dell’Hb A2 in presenza di difetti β-globinici e la relativa interpretazione. Ma nella pratica quotidiana si osservano casi con valori di Hb A2 sensibilmente più bassi rispetto a quelli di riferimento. È importante inoltre ricordare che la corretta interpretazione della misura dell’Hb A2 e del suo possibile significato clinico non può mai prescindere dalla conoscenza di altri parametri analitici fondamentali forniti dall’emocromo e dall’assetto marziale, come indicato dalle suddette raccomandazioni. La presenza di un valore ridotto di Hb A2 può rappresentare un utile indicatore per segnalare la presenza di difetti globinici e non solo, per cui in molti casi richiede ulteriori indagini biochimiche o molecolari.
Cause di Riduzione dell'HbA2 e Loro Implicazioni
I quadri che di seguito vengono riportati rappresentano le circostanze più frequentemente associate a valori ridotti di HbA2 in soggetti adulti non trasfusi. La variabilità dell’Hb A2 nel formare il tetramero (α2 δ2) può anche essere in relazione alla competizione e alla conseguente diversa affinità reciproca di tutte le catene prodotte nel soggetto adulto (α, β e δ normali o mutate), così come accade per le varianti Hb in generale.
Carenza di Ferro e HbA2
La sideropenia è una condizione dovuta a diverse cause ed è molto diffusa nella popolazione. Essa può essere associata ad anemia (anemia sideropenica) quando la carenza di ferro è particolarmente marcata e si prolunga nel tempo. Secondo l’OMS, lo stato di anemia si intende per valori di Hb nel sangue inferiori a 120 g/L nella donna e 134 g/L nell’uomo.
Una lieve carenza di ferro modifica poco l’MCV e l’Hb A2, ma quando subentra l’anemia, i valori di questi parametri possono diminuire in modo significativo, in particolare l’Hb A2 può arrivare anche al di sotto del 2%. L’anemia sideropenica riduce anche i valori di Hb A2 in presenza di difetti talassemici; nel caso di β0 talassemia, però, ciò non avviene in modo significativamente marcato e tale da compromettere la diagnosi del portatore classico. L’anemia sideropenica potrebbe tuttavia condizionare negativamente l’interpretazione del valore dell’Hb A2 quando sono presenti difetti β+ o β++. Pertanto, le linee guida prevedono in ogni caso la ripetizione degli esami dopo correzione del bilancio marziale.
Alfa-Talassemia e HbA2
Non vi è un parametro eritrocitario o emoglobinico che può definire un tratto α-talassemico, così come accade invece per le forme classiche della β-talassemia. L'informazione genetica per le catene α è contenuta in 4 geni: nel caso dell'α-talassemia perciò il difetto può derivare dall'alterazione di tutte e quattro i geni (condizione non compatibile con lo sviluppo del feto), di tre geni (presenza di Hb formata da complessi di quattro catene ß chiamata HbH, accompagnata da anemia emolitica, splenomegalia ecc.), di due geni (tratto talassemico con presenza di anemia, globuli rossi di volume ridotto, ma con percentuali di HbA2 e HbF di solito normali), di un gene (condizione indistinguibile dalla normalità, ma con possibile trasmissione del carattere alla prole).
L’Hb A2 nel portatore di α-talassemia si presenta variabilmente e lievemente ridotta, mentre l’MCV risulta diminuito in modo più significativo solo nelle forme con due geni α non funzionanti (α0 talassemia (--/αα) o nell'α+ omozigote (-α/-α)). Si può osservare pertanto che l’Hb A2, nel portatore di α-talassemia, si presenta con valori che di solito rientrano tra quelli più bassi dell’intervallo di normalità. Quando però queste forme di α-talassemia si associano alla carenza di ferro, lo scostamento dalla norma dei valori dell’Hb A2 e dell’MCV diventa significativo. Ma, in caso di emoglobinosi H, e cioè quando tre dei quattro geni α non funzionano (--/-α), i valori dell’Hb A2 sono di solito inferiori a 1,5%, come ampiamente documentato nel “caso 3” di questo portale.
Varianti delle Catene Delta o Delta-Talassemie e HbA2
Quando uno dei due geni δ non consente la sintesi di catene utili alla formazione di Hb A2 (delta talassemia) o le produce mutate (varianti delta, α2 δ2X), si avrà un valore di Hb A2 ridotto fino al 50% rispetto alla norma. Le varianti delta possono essere quantificate dai sistemi separativi utilizzati, oppure potrebbero essere non quantificabili perché co-migranti con Hb A o altre varianti eventualmente presenti. Talvolta l’Hb A2 potrebbe essere invece sintetizzata in misura tanto ridotta da risultare in quantità inferiore alla sensibilità della strumentazione utilizzata. Nel caso di δ-talassemia, si osserva sempre e solo una riduzione dell’Hb A2 pari a circa il 50% rispetto al valore di riferimento normale. Nei rari casi in cui i geni δ presentano difetti in omozigosi (δ-talassemia omozigote), l’Hb A2 risulterà assente. Di seguito viene riportato un esempio in eterozigosi con alcuni parametri in tabella (paziente 1) ed il profilo elettroforetico (figura 1) che mostra la separazione della variante Hb A2-Fitzroy.
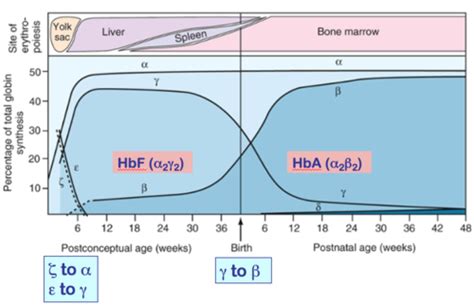
Varianti delle Catene Alfa e HbA2
Altra circostanza nella quale possiamo riscontrare un valore sensibilmente ridotto dell’Hb A2 è la presenza di una variante delle catene α globiniche. Ciò è dovuto al fatto che anche le catene α contribuiscono a formare l’Hb A2 e quindi il tetramero globinico risentirà della presenza di tale mutazione (α2Xδ2). Vi sono circostanze in cui non è possibile osservare l’Hb A2 mutata (A2-X); ciò può accadere quando la variante α co-migra con l’Hb A o quando la variante è particolarmente instabile. Di converso, occorre sottolineare che una eventuale contemporanea presenza di α-talassemia, così come contribuisce ad incrementare la quantità relativa della variante α, incrementerà anche il valore relativo di Hb A2-X, riducendo quello dell’Hb A2 normale. Viene qui riportato un esempio con alcuni parametri in tabella (paziente 2) ed il profilo elettroforetico (figura 2) da cui risulta la presenza dell’Hb J Rovigo (variante α) e dell’Hb A2 Rovigo secondaria alla variante α.
Emoglobinopatie e Talassemie: Quadro Generale e Varianti Specifiche
Un’emoglobinopatia è un disordine ematico ereditario caratterizzato dalla presenza di forme anomale dell’emoglobina (varianti emoglobiniche) o dalla riduzione della produzione della stessa (talassemia). La presenza di mutazioni nei geni codificanti per le catene globiniche può determinare la produzione di emoglobine anomale e quindi la presenza di emoglobinopatie. Attualmente sono state rilevati circa 1000 tipi di emoglobinopatie.
Le mutazioni possono comportare la produzione di catene globiniche strutturalmente alterate o anche la perdita della produzione di uno o più tipi di catene globiniche. Possono inoltre influenzare la struttura dell’emoglobina, la sua funzionalità, la sua velocità di produzione o anche la sua stabilità. Circa il 7% della popolazione mondiale è portatore in eterozigosi di una variante genetica in una delle catene dell’emoglobina; il tasso di mutazione può variare notevolmente in base all’etnia.
La talassemia, invece, è causata dalla diminuita produzione o carenza di una delle catene globiniche. Le talassemie sono causate da un difetto ereditario che impedisce la normale sintesi delle catene α (α-talassemia) o delle catene ß (ß-talassemia). L'informazione genetica per le catene ß è contenuta in 2 geni: nel caso della ß-talassemia il difetto può derivare dall'alterazione di entrambi i geni (forma omozigote), e in questo caso la malattia è definita ß-talassemia major o morbo di Cooley (si manifesta subito dopo la nascita con una anemia molto grave, che necessita di trasfusioni periodiche di sangue), oppure dall'alterazione di un solo gene (forma eterozigote), e in questo caso il difetto viene chiamato ß-talassemia minor o tratto talassemico (la maggior parte delle persone con talassemia minor non presenta sintomi; i globuli rossi nel loro sangue sono in numero maggiore che nel normale, ma di volume ridotto - da cui il termine di microcitemia - e poveri di emoglobina).
Esistono molteplici varianti emoglobiniche. Alcune silenti (ossia senza segni e sintomi evidenti) ed altre in grado di influenzare la funzionalità e/o la stabilità della molecola emoglobinica. È possibile che la stessa persona erediti due differenti geni anomali, uno da ciascun genitore, con conseguente combinazione delle varianti emoglobiniche rilevate dai test. Questa condizione è nota come eterozigosi composta o doppia eterozigosi. Spesso, le forme meno comuni prendono il nome dal luogo di appartenenza della/e famiglia/e in cui la variante genetica è stata identificata per la prima volta.
Le principali varianti emoglobiniche includono:
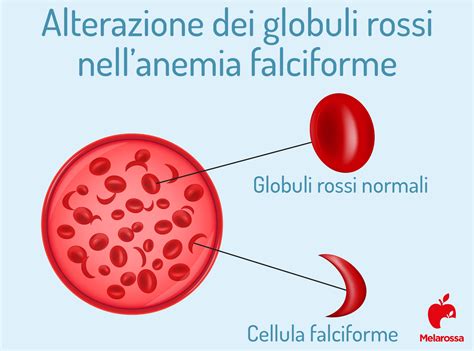
- Emoglobina S (HbS): responsabile dell’anemia falciforme. Si tratta di una patologia a frequenza molto variabile, ma comunque maggiormente presente nelle aree nelle quali la malaria è o è stata endemica, a causa di un vantaggio evolutivo dei portatori nei confronti della malaria. Le persone affette da anemia falciforme presentano entrambe le copie del gene responsabile della produzione di HbS, e pertanto presentano elevate quantità di HbS. Le persone con “tratto” falciforme (eterozigoti) presentano circa il 40% di HbS e il 60% di HbA normale. La presenza di HbS determina, in presenza di basse concentrazioni di ossigeno (come può accadere durante un esercizio fisico o in caso di infezioni polmonari), la variazione della forma dei globuli rossi che assumono la caratteristica forma a falce. Gli eritrociti falciformi possono bloccare i piccoli vasi, causando episodi dolorosi, scompensi circolatori e diminuzione dell’apporto di ossigeno ai tessuti con diminuzione della sopravvivenza cellulare.
- Emoglobina C (HbC): lo stato di portatore di HbC (eterozigoti) è presente in circa il 2-3% delle persone di origine africana. Lo stato di omozigosi comunque è un evento raro ed è responsabile di effetti relativamente lievi.
- Emoglobina E (HbE): è una delle varianti beta-globiniche più comuni nel mondo, con una maggiore prevalenza nelle persone originarie del sud-est asiatico. I soggetti omozigoti per HbE in genere hanno una lieve anemia emolitica, macrocitosi ed un moderato ingrossamento della milza.
- Emoglobina H (HbH): si forma in alcuni casi di α-talassemia. È composta da quattro catene beta globiniche (β4) e viene prodotta in caso di grave carenza delle catene alfa.
- Emoglobina di Bart: si sviluppa nei feti affetti da α-talassemia. È composta da quattro catene globiniche gamma (γ4) e viene prodotta in caso di grave carenza delle catene alfa in maniera analoga a quanto avviene per l’HbH.
Anemia in Gravidanza: Rilevanza Clinica e Gestione
I quadri di anemia ed ematocrito basso in gravidanza sono frequenti e possono preoccupare la mamma in attesa. In tutto questo non bisogna mai dimenticare che il feto in crescita assorbe numerosi nutrienti dal corpo della madre e che tra questi è presente il ferro. Alla luce di ciò, una blanda riduzione dei livelli di emoglobina va considerata come fisiologica (emodiluizione fisiologica). L'emodiluizione in gravidanza va monitorata.
L'emoglobina è una proteina presente nei globuli rossi e avente il compito di trasporto dell'ossigeno. Affinché il midollo osseo riesca a sintetizzarla, è essenziale il ferro, che si accumula nel fegato sotto forma di ferritina, proteina i cui livelli vanno monitorati tramite gli esami del sangue in gravidanza. L’ematocrito è la percentuale, su tutto il sangue, di parte corpuscolata (alla sua composizione contribuiscono, come già accennato, soprattutto i globuli rossi). Nella donna sana non gravida, i valori normali vanno dal 38 al 45%. In caso di gravidanza quasi a termine fisiologica e singola, si può arrivare al 34% e al 30% nell’eventualità di una gravidanza multipla (parlando sempre di valore normale a fine gestazione).
Per poter parlare in maniera specifica di anemia ed ematocrito basso in gravidanza, i valori devono essere i seguenti:
- 1° trimestre: emoglobina sotto gli 11 g/dl ed ematocrito inferiore al 33%;
- 2° trimestre: valori dell’emoglobina inferiori ai 10,5 g/dl ed ematocrito inferiore al 32%;
- 3° trimestre: emoglobina pari a meno di 11 g/dl ed ematocrito inferiore al 33%.

L'esecuzione del prelievo per l'emocromo è indicata all’inizio della gravidanza (idealmente entro le prime tredici settimane) e dovrà poi essere ripetuta a 28 settimane, in modo da disporre di un tempo adeguato per il trattamento, qualora necessario. Con l’emocromo si individuano i casi con livelli di emoglobina bassa, ossia inferiori allo standard di riferimento per l’epoca di gravidanza. L'esame dell'emocromo va effettuato a inizio gestazione, se possibile non oltre la 13° settimana, a metà della gravidanza e verso la fine. L’esecuzione attorno alla ventesima settimana ha un ruolo di particolare rilevanza. Questo perché, più o meno a metà gravidanza, viene toccato il cosiddetto nadir, il livello più basso dell’emoglobina durante la gestazione. In tale fase, è possibile capire se l’emodiluizione è fisiologica oppure se si ha a che fare con una situazione di anemia ed ematocrito basso in gravidanza, con ovvia necessità di integrare il ferro.
Quando si parla di anemia ed ematocrito basso in gravidanza si inquadra, come già accennato, una situazione che ha alla base un’emodiluizione quasi sempre fisiologica. Tuttavia, possono esserci anche altre cause. Ecco quali:
- Carenza di ferro;
- Carenza di folato, evenienza che riguarda lo 0,5 - 15% delle gravide e che espone a un maggior rischio di malformazioni al tubo neurale del nascituro e di sindrome feto-alcolica;
- Carenza di vitamina B12, nutriente decisivo per la sintesi dei globuli rossi.
Quando i valori dei globuli rossi sono bassi in gravidanza, la mamma in attesa può avere a che fare con sintomi quali mal di testa, pallore, un generale senso di affanno, l’ipotensione, per non parlare dell’incremento anomalo della frequenza cardiaca. Si tratta di quadri meritevoli di immediata attenzione medica, onde evitare un impatto negativo sullo sviluppo del feto e sulle sue riserve di ferro alla nascita.

Se non si procede tempestivamente a trattare la situazione a fronte di valori inferiori agli 8,5 g/dl, aumenta il rischio di parto pretermine, basso peso del feto alla nascita, sviluppo di ritardi psicomotori da parte del bimbo nel corso della crescita. La mamma, invece, è fortemente esposta al rischio di gestosi (o preeclampsia), di distacco di placenta e di emorragia nel post-parto. In assenza di anemia falciforme o talassemie, il trattamento dell’anemia in gravidanza prevede la somministrazione di ferro per bocca al fine di ristabilire le riserve di questo elemento. In presenza di emoglobina inferiore a 11 g/dl a inizio gravidanza, si procede con un trattamento di profilassi. Lo step in questione è necessario in quanto, con il successivo aumentare dell’emodiluizione fisiologica, si può andare sotto i 10 mg/dl.
ANEMIA IN GRAVIDANZA: i miei consigli
Diagnosi e Screening delle Emoglobinopatie: Il Ruolo dell'Assetto Emoglobinico
Per la diagnosi delle emoglobinopatie l’esecuzione di un singolo test non è sufficiente. Questa richiede la valutazione complessiva di una serie di esami, eseguiti secondo processi ben definiti. I test di primo livello per la ricerca delle emoglobinopatie in genere utilizzano metodiche volte alla determinazione del tipo e della quantità di emoglobine presenti nel sangue del paziente in esame (assetto emoglobinico). L’assetto emoglobinico si prefigge di rilevare eventuali varianti emoglobiniche e/o la quantità relativa dei diversi tipi di Hb. La maggior parte delle varianti emoglobiniche più comuni o delle talassemie possono essere identificate utilizzando una combinazione di questi test. La rilevazione della quantità relativa della variante emoglobinica presente è un valido ausilio diagnostico.
Test molecolari ricercano le mutazioni presenti nei geni codificanti per le catene globiniche alfa e beta. Il test per la ricerca delle emoglobinopatie viene richiesto come parte dei programmi di screening neonatale (attualmente attivo solo in alcune regioni in Italia). L’esame può essere richiesto nel caso in cui si sospetti un’emoglobinopatia in presenza di segni e sintomi caratteristici. Lo screening delle emoglobinopatie consente di identificare e quindi potenzialmente anche trattare i neonati con disordini congeniti entro pochi giorni dalla nascita. In questo modo possono essere evitati problemi di salute potenzialmente letali o anche responsabili di disabilità permanenti. Questo esame non viene eseguito in tutti i laboratori.
Nella valutazione delle emoglobinopatie deve essere prestata molta attenzione all’interpretazione dei risultati. I risultati in genere riportano il tipo di Hb presenti (quando identificabile) e la loro quantità relativa. Le trasfusioni di sangue possono interferire con la valutazione delle emoglobinopatie, poiché il metodo analitico non è in grado di distinguere tra l’emoglobina del donatore e quella del paziente, interferendo potenzialmente con i risultati del test. Un paziente dovrebbe aspettare diversi mesi dopo una trasfusione prima di sottoporsi a questo esame.
Approfondimenti Diagnostici e Raccomandazioni per l'HbA2
I valori di Hb A2 inferiori a 2,5% devono essere valutati considerando le possibili cause di decremento sopra esposte. Tuttavia, altre cause più rare possono essere presenti. Per queste si rimanda alle raccomandazioni per la diagnostica di 1° livello (1). È opportuno ribadire che l’intervallo di normalità dell’Hb A2 indicato dalle citate raccomandazioni rappresenta l’insieme dei valori normali riscontrabili nel 95% della popolazione. Tuttavia, così come si conoscono valori normalità «borderline alti», che occorre tener presente, confermare e caratterizzare nel corso degli esami di prevenzione della beta talassemia, esistono valori «borderline bassi» che possono rientrare nella normalità ma senza presentare significato clinico.
Nella pratica di laboratorio occorre considerare che:
- Valori bassi (da 2,0 a 2,5%) di Hb A2 in assenza di microcitosi, carenza marziale o anemia, non hanno mai significato clinico.
- Osservando o sospettando lo sdoppiamento dell’Hb A2 (dovuto alla presenza di varianti δ o α), occorre sempre verificare che la somma (Hb A2 + Hb A2-X) risulti sempre inferiore a 3,2%, con o senza microcitosi. Questa operazione è importante per poter riconoscere l’aumento relativo delle catene δ ed evitare il rischio di non diagnosticare la presenza di una eventuale β talassemia.
- La presenza di Hb A2 inferiore a 1,5-1,6%, in condizioni di sideremia normale e in assenza di microcitosi, anemia e varianti deve far sospettare il coinvolgimento di un difetto dei geni δ. In questi casi l’approfondimento molecolare non è ritenuto essenziale ma opzionale.
- Valori dell’Hb A2 inferiori a 2,4% associati ad anemia e microcitosi, devono sempre essere valutati alla luce dell’assetto marziale e possono prevedere approfondimenti molecolari per escludere difetti α-talassemici, soprattutto in gravidanza o in età fertile.
Il trattamento di alcuni tipi di emoglobinopatie può comportare l’utilizzo di terapie di supporto, ad esempio durante la comparsa di crisi in corso di anemia falciforme. L’obiettivo è quello di alleviare il dolore e di minimizzare le complicanze. Talvolta, in caso di anemia grave, è necessario ricorrere a trasfusioni di sangue. Meno frequentemente, possono essere utilizzati anche altri trattamenti. In utero, il feto normale produce una piccola quota di emoglobina adulta (2,5-5%). Il feto con talassemia maior ne produce ancor meno (inferiore al 2%). Per rilevare durante la gravidanza se un feto è affetto da talassemia maior, è possibile determinare la quantità di emoglobina adulta presente in un campione di sangue prelevato mediante cordocentesi.
ANEMIA IN GRAVIDANZA: i miei consigli
tags: #assetto #emoglobinico #gravidanza