L'anticipazione genetica rappresenta un fenomeno biologico di fondamentale importanza nella comprensione della trasmissione di alcune malattie ereditarie, in particolare quelle legate alle espansioni di triplette nucleotidiche. Questo meccanismo, che porta a manifestazioni sempre più severe e ad un'età di insorgenza precoce nelle generazioni successive, è di cruciale rilevanza per i pazienti, le loro famiglie e i professionisti della salute.
Il Concetto di Anticipazione Genetica
L'anticipazione è un fenomeno genetico per cui quando un disturbo genetico viene trasmesso alla generazione successiva, i sintomi del disturbo genetico diventano evidenti in età precoce con ogni generazione. Nella maggior parte dei casi, si nota anche un aumento della gravità dei sintomi. In genetica, fenomeno per il quale nel passaggio alle generazioni successive diminuisce l’età di insorgenza o aumenta la gravità di una malattia. È un aspetto complesso e delicato nella gestione di patologie come la Distrofia Miotonica di tipo 1, spesso poco conosciuto tra i pazienti e le loro famiglie.
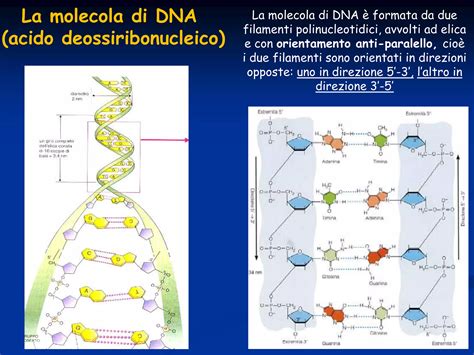
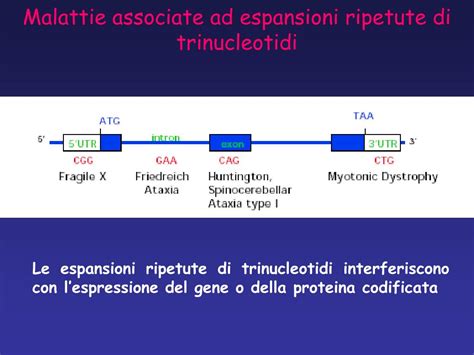
Questo meccanismo spiega perché una malattia ereditaria possa manifestarsi in forme progressivamente più severe passando da una generazione all’altra. L’anticipazione è determinata da mutazioni, dette dinamiche, dovute all’espansione di ripetizioni di trinucleotidi (TNRE, trinucleotide repeat expansion) come CAG, CTG, CGG o GAA. Queste espansioni causano varie malattie ereditarie, quali la sindrome da X-fragile, l’atrofia spinobulbare, la distrofia miotonica, l’atassia di Friedreich. La gravità o l’età di insorgenza di queste patologie sono correlate alla lunghezza delle unità ripetute e la lunghezza delle ripetizioni tende a crescere man mano che il gene viene trasmesso alle generazioni successive. Inoltre, la quantità di ripetizioni è diversa a seconda che la malattia sia ereditata dalla madre o dal padre.
Cromosomi e alleli
Basi Molecolari dell'Anticipazione Genetica
L'anticipazione è comune nei disturbi da ripetizione delle triplette di nucleotidi, in cui si verifica una mutazione dinamica nel DNA. Le ripetizioni del trinucleotide sono evidenti in numerosi loci del genoma umano. Sono stati trovati in introni, esoni e UTR 5 'o 3'. Sono costituiti da un modello di tre nucleotidi (ad esempio CGG) che viene ripetuto più volte. Il fenomeno dell'anticipazione è sostanzialmente dovuto a sequenze ripetitive instabili le quali possono andare incontro a espansione durante la meiosi, aumentando nella progenie il numero di ripetizioni delle triplette nel gene responsabile della patologia.
L’espansione delle sequenze ripetute può alterare sia l’espressione sia la struttura di un gene particolare a seconda che si trovino all’interno del gene (negli esoni o negli introni) o nelle sequenze alle due estremità (5’ o 3’) di esso. Il meccanismo alla base dell'espansione delle ripetizioni di terzine non è ben compreso. Per molti loci, l'espansione del trinucleotide è innocua, ma in alcune aree l'espansione ha effetti dannosi che causano sintomi.
Quando la ripetizione della tripletta di nucleotidi è presente all'interno della regione codificante per la proteina, l'espansione della ripetizione porta alla produzione di una proteina mutante con aumento di funzionalità (gain of function). Questo è il caso della malattia di Huntington, in cui la ripetizione del trinucleotide codifica un lungo tratto di residui di glutammina. Quando la ripetizione è presente in una regione non tradotta, potrebbe influire sull'espressione del gene in cui si trova la ripetizione (es. Fragile X) o su molti geni attraverso un effetto negativo dominante. Per avere un effetto deleterio, il numero di ripetizioni deve superare una certa soglia.
La Distrofia Miotonica di Tipo 1 come Modello di Anticipazione
La Distrofia Miotonica di tipo 1 (DM1) rappresenta una malattia rara di natura ereditaria, caratterizzata da una trasmissione autosomica dominante con penetranza pressoché completa. La condizione origina dall’espansione patologica delle ripetizioni di una tripletta di basi CTG, localizzata in una porzione non trascritta del gene DMPK sul cromosoma 19. Quando queste ripetizioni superano quota cinquanta, si manifesta il quadro patologico.
Ad esempio, gli individui normali hanno tra le 5 e le 30 ripetizioni CTG entro il 3 'UTR di DMPK, il gene che viene alterato nella distrofia miotonica. Se il numero di ripetizioni è compreso tra 50 e 100, la persona è solo lievemente colpita, forse avendo solo la cataratta. Tuttavia, l'instabilità meiotica potrebbe comportare una mutazione dinamica che aumenta il numero di ripetizioni nella prole che eredita l'allele mutante. Una volta che il numero di copie raggiunge oltre 100, la malattia si manifesterà prima nella vita e i sintomi saranno più gravi, inclusa la miotonia elettrica.
Il grado di espansione della tripletta CTG risulta strettamente correlato alla gravità del fenotipo clinico e all’età in cui compaiono i primi sintomi. Espansioni comprese tra trentacinque e cinquanta triplette rimangono generalmente asintomatiche, mentre quelle tra cinquanta e centocinquanta si associano più frequentemente a forme oligosintomatiche con manifestazioni cliniche limitate. Questo meccanismo molecolare si accompagna a un fenomeno definito anticipazione genetica, attraverso il quale l’espansione patologica e di conseguenza la gravità del fenotipo tendono ad aumentare progressivamente di generazione in generazione all’interno dello stesso nucleo familiare.
Frequentemente accade che la presenza della malattia in un genitore asintomatico o affetto dalla condizione in forma lieve e non riconosciuta venga scoperta proprio attraverso la nascita di un bambino con distrofia miotonica di tipo 1 o nella sua forma congenita.

Altre Malattie con Anticipazione Genetica
L'anticipazione è comune nei disturbi da ripetizione delle triplette di nucleotidi, come la malattia di Huntington e la distrofia miotonica. Tutte queste malattie hanno in comune sintomi neurologici.
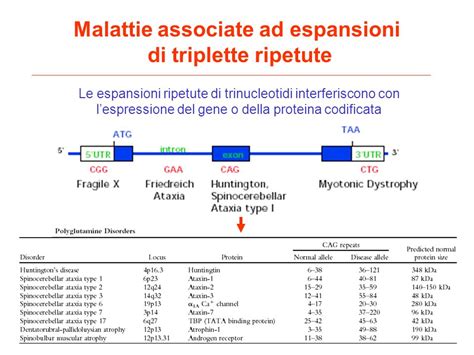
Nelle malattie umane corea di Huntington, DRPLA (dentatorubral-pallidoluysian atrophy), atrofia spinobulbare, atassia spinocerebellare tipo 1, atassia spinocerebellare tipo 3, la ripetizione CAG (citosina, adenina, guanina) è localizzata nella regione codificante del primo esone del gene. Queste ripetizioni vengono tradotte e compaiono nella proteina come una sequenza di glutammine, cioè dell’amminoacido specificato dalla tripletta CAG. Tutte le malattie causate da ripetizioni CAG implicano una menomazione a livello neuronale che si sviluppa tardivamente e sono ereditate con modalità autosomica dominante. Dato che le diverse proteine interessate nelle malattie sopra citate non presentano omologie fra loro e svolgono differenti funzioni nella cellula, si ipotizza che proprio la glutammina in eccesso conferisca il fenotipo mutante.
Nelle sindromi da X-fragile (FRAXA e FRAXE), invece, le ripetizioni della tripletta CGG (citosina, guanina, guanina) sono localizzate all’estremità 5’ non codificante del primo esone del gene.
Il fenomeno dell'anticipazione si verifica spesso nella trasmissione paterna della Malattia di Huntington: solitamente un esordio giovanile è subordinato alla presenza di almeno 60 triplette CAG in porzioni codificanti del gene.
Counselling Genetico e Pianificazione Familiare
Le problematiche derivanti da questa realtà biologica sono molteplici e spesso sottovalutate. Risulta quindi essenziale che la diagnosi genetica della condizione, sia negli uomini che nelle donne in età fertile o con premutazione, venga accompagnata da un counselling genetico adeguato e approfondito.
Il paziente o i genitori di un soggetto affetto necessitano di essere informati in modo chiaro sulla trasmissibilità autosomica dominante della malattia, sul fenomeno dell’anticipazione genetica e sul rischio specifico della forma congenita tra la prole delle donne affette. Un percorso di pianificazione familiare consapevole dovrebbe includere la discussione approfondita delle possibilità offerte dalla diagnosi prenatale, dall’interruzione volontaria di gravidanza e dalle tecniche di procreazione medicalmente assistita.
Il riconoscimento precoce della malattia nei pazienti affetti da distrofia miotonica di tipo 1 da parte dei Medici di Medicina Generale e il conseguente invio tempestivo presso centri specializzati può contribuire in modo sostanziale ad affrontare adeguatamente la complessa problematica della procreazione e della gravidanza in questa patologia.
Gravidanza e Gestione Clinica nella Distrofia Miotonica
Quando la gravidanza è già in corso, diventa fondamentale considerare tutte le problematiche possibili associate alla condizione materna. Tra queste si annoverano il rischio aumentato di gravidanza ectopica, parto prematuro, emorragia post-partum, sviluppo di diabete gestazionale e possibile aggravamento della debolezza muscolare.
La gestione del parto richiede necessariamente un ambiente ospedaliero attrezzato, con la disponibilità di personale ostetrico e pediatrico esperto nelle manovre di rianimazione neonatale. La presenza di professionisti preparati a gestire eventuali complicanze rappresenta un elemento determinante per la sicurezza sia della madre che del neonato.

Diagnosi Genetica Preimpianto (PGT)
La Diagnosi Genetica Pre-Impianto (DGP) o Test Genetico Pre-Impianto (PGT), è l’analisi della genetica dell’embrione mediante lo studio di una biopsia delle sue cellule prima di essere trasferito nell’utero materno. È un insieme di tecniche che permettono di individuare la presenza di anomalie cromosomiche e/o patologie genetiche nei gameti (ovociti e spermatozoi), o negli embrioni, prima che vengano trasferiti nell’utero. L’obiettivo è identificare e prevenire durante la fase embrionale le malattie gravi causate da alterazioni genetiche. Le tecniche di diagnosi genetica pre-impianto rappresentano una valida alternativa alla diagnosi prenatale in coppie con aumentato rischio di trasmettere malattie genetiche ai loro futuri nascituri. Tuttavia è da tener presente che si tratta di una procedura complessa, invasiva e non priva di rischi.
La PGT è indicata nelle coppie affette o portatrici di una malattia genetica che vogliono avere un figlio sano per quella malattia. Per poter eseguire la PGT è necessario avere lo studio genetico nei futuri genitori. Quindi, il primo passo è quello di eseguire lo studio genetico, cioè identificare il difetto nel gene (mutazione) che sta causando la malattia. Una volta che le fasi precedenti della DGP sono state completate (studio genetico e informatizzazione) possiamo iniziare il ciclo di DGP con garanzie. Per farlo, la coppia deve sottoporsi al trattamento di fecondazione in vitro (FIVET). Dobbiamo aspettare che gli embrioni risultanti dal processo si siano divisi per estrarre diverse cellule dall'embrione (biopsia embrionale). Le biopsie saranno analizzate nel laboratorio di biologia molecolare per sapere se gli embrioni biopsiati sono sani o meno per la malattia studiata.
PGT per Malattie Monogeniche (PGT-M)
La PGT-M è applicabile quando uno o entrambi gli aspiranti genitori sono portatori di o affetti da una patologia genetica potenzialmente trasmissibile alla prole. Consiste nell’analisi genetica degli embrioni di una coppia portatrice di una malattia ereditaria. Permette di rilevare l’alterazione o la mutazione in un gene che causa la malattia. Il primo passo consiste nell’effettuare uno studio genetico dei futuri genitori per individuare l’errore nel gene (mutazione) che causa la malattia - studio genetico-. Una volta che abbiamo lo studio genetico, il passo successivo è quello di effettuare lo studio informativo che ci permette di stabilire una strategia diagnostica specifica per questa malattia in quella famiglia. Le alterazioni possono essere autosomiche recessive, autosomiche dominanti e legate al cromosoma X come la sindrome di X fragile, la emofilia A, la fibrosi cistica, la malattia di Huntington, l’anemia falciforme, la malattia di Marfan, ecc.
Per procedere con il trattamento di analisi PGT-M è necessario effettuare uno studio preliminare sul DNA degli aspiranti genitori e, in alcuni casi, anche di altri membri della famiglia. Questo studio di DNA viene fatto analizzando campioni biologici, come sangue, saliva o liquido seminale e serve per rendere più accurata la successiva diagnosi. Una volta completata questa procedura preliminare, si esegue un ciclo di procreazione assistita con tecnica ICSI e gli embrioni ottenuti sono sottoposti a biopsia, cioè un prelievo di materiale genetico assolutamente innocuo. Dal materiale prelevato viene estratto DNA che viene amplificato per ottenerne più copie e verificare se è presente la mutazione associata alla patologia. L’accuratezza della tecnica di analisi PGT-M è del 95-98%. Ad oggi esistono protocolli diagnostici per oltre 120 malattie monogeniche, autosomiche dominanti, recessive o legate al cromosoma X.
PGT per Anomalie Cromosomiche (PGT-A)
La PGT-A consente di analizzare l’assetto cromosomico degli embrioni per individuare eventuali anomalie nel numero di cromosomi. Nel momento in cui si uniscono il patrimonio genetico della madre e quello del padre, è possibile che avvengano degli errori: si possono quindi avere copie di cromosomi in più o in meno. L’anomalia cromosomica più nota è sicuramente la Sindrome di Down, causata dalla presenza di tre copie del cromosoma 21. Si esegue un ciclo di procreazione assistita con tecnica ICSI e gli embrioni ottenuti sono sottoposti a biopsia, cioè un prelievo di materiale genetico assolutamente innocuo. Dal materiale prelevato viene estratto DNA che viene amplificato per ottenerne più copie e verificare se sono presenti anomalie. L’accuratezza della tecnica di analisi PGT-A è del 95-98%. Questo progresso fornisce informazioni aggiuntive cruciali. Il PGT-A Advanced permette anche di conoscere l’origine specifica di queste alterazioni per selezionare gli embrioni con la maggiore probabilità di generare una gravidanza.
La PGD per lo studio delle alterazioni cromosomiche (PGD), presenta qualche dibattito sulla bontà che la sua applicazione potrebbe avere sul miglioramento della tassa di successo delle tecniche di riproduzione assistita. Solo gli embrioni che non hanno un numero alterato di cromosomi daranno origine a un bambino sano. Ci sono alterazioni cromosomiche che sono incompatibili con la vita e che impediscono all'embrione di svilupparsi nelle sue prime fasi e persino di impiantarsi nell'utero materno. Tra le alterazioni cromosomiche ce ne sono alcune meno dannose per l'embrione che gli permettono di impiantarsi, ma che impediscono alla gravidanza di svilupparsi correttamente e possono finire in un aborto spontaneo, o che il futuro bambino soffra di sindromi diverse come la sindrome di Down, Patau o Edwards. Usando la PGS evitiamo il trasferimento di embrioni che non daranno mai origine a un bambino sano, perché saranno scartati. L'uso della PGS permette di ridurre l'incertezza nei pazienti. Da un lato, garantisce che il loro embrione è sano e che la tecnologia più avanzata è stata applicata per assicurarlo.
Cromosomi e alleli
PGT per Riarrangiamenti Strutturali (PGT-SR)
Per la PGT-SR l’indicazione è che uno o entrambi gli aspiranti genitori siano portatori di una traslocazione. Parliamo di traslocazione reciproca bilanciata quando la riorganizzazione non produce perdita né acquisizione di materiale genetico. Una parte di un cromosoma si è inserita in una posizione insolita all’interno dello stesso o di un altro cromosoma. Parliamo di inversioni quando una parte del cromosoma si rompe in due punti e il frammento interno si inverte al contrario per poi riunirsi.
Per procedere con il trattamento di analisi PGT-SR è necessario effettuare uno studio preliminare sul DNA degli aspiranti genitori e, in alcuni casi, anche di altri membri della famiglia. Questo studio di DNA viene fatto analizzando campioni biologici, come sangue, saliva o liquido seminale e serve per rendere più accurata la successiva diagnosi. Una volta completata questa procedura preliminare, si esegue un ciclo di procreazione assistita con tecnica ICSI e gli embrioni ottenuti sono sottoposti a biopsia, cioè un prelievo di materiale genetico assolutamente innocuo. Dal materiale prelevato viene estratto DNA che viene amplificato per ottenerne più copie e verificare se è presente la traslocazione oggetto di studio.
Metodologie di Biopsia Embrionale
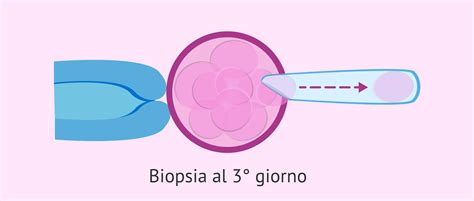
Entrambe le metodiche sono precedute dalla fecondazione in vitro (effettuata mediante FIVET o ICSI), seguita dalla biopsia degli embrioni così generati. La metodica più utilizzata fino a pochi anni fa prevedeva la biopsia del materiale da esaminare al terzo giorno di sviluppo embrionale, con il prelievo di una o due cellule (blastomeri) dall’embrione. Il risultato dell’analisi può essere ottenuto entro 24h per cui in caso di assenza di anomalie genetiche, il transfer si può effettuare in quinta giornata (D5) allo stadio di blastocisti. Il limite è rappresentato dal fatto che l’embrione risente del mosaicismo, ossia di un differente stato genetico che varia da blastomero a blastomero, comportando l’esclusione di embrioni in cui difetti genetici erano presenti solo nel blastomero analizzato. Inoltre le aneuploidie potrebbero essere corrette durante l’evoluzione da embrione a blastocisti.
Negli anni più recenti, l’ottimizzazione delle condizioni di coltura ha permesse il prelievo bioptico al quinto giorno di sviluppo embrionale, cioè allo stadio di blastocisti. L’analisi viene effettuata principalmente sulle cellule del trofoectoderma (cellule embrionali che contribuiranno successivamente alla formazione della placenta) oppure sul fluido del blastocele (liquido contenuto all’interno della blastocisti). L’analisi del materiale trofoectodermico permette di studiare e valutare problemi genetici in più cellule, alleviando il problema del mosaicismo. Quest’ultima tecnica è stata sviluppata proprio da S.I.S.Me.R., e ha il vantaggio di una minore invasività perché non prevede il prelievo di cellule. Questo consente l’aspirazione di cellule dal trofoectoderma, in questo modo la massa delle cellule interne, che darà origine al feto nelle fasi di sviluppo successive, non è danneggiata, garantendo un trauma minore per l’embrione rispetto al prelievo in terza giornata.
Il Mosaicismo Embrionale
È ampiamente noto che gli embrioni umani presentano un certo grado di mosaicismo, tuttavia, era difficile diagnosticarlo. Oggi, grazie allo sviluppo delle tecniche di analisi genetica, possiamo sapere se c’è un insieme di cellule normali ed alterate (mosaico) nell’embrione. Quello che resta da determinare è se questo fatto influisce in qualche modo sull’embrione. La PGS analizza la parte esterna dell’embrione con l’obiettivo di lasciare intatta quella parte (massa cellulare interna) che darà origine al bambino, perché ci sono studi scientifici che hanno dimostrato un alto grado di correlazione tra i due. Non è stato dimostrato se altera in qualche modo l’embrione.
Vantaggi e Considerazioni della PGT
Evita il trasferimento di embrioni che non anniderebbero. Si riduce il tempo necessario per ottenere una gravidanza. Costo inferiore. Migliora il benessere psicologico. Il PGD applicata allo studio delle malattie genetiche che colpiscono un gene, presenta un chiaro beneficio che non solleva alcun dibattito sulla sua utilità. Tuttavia, la PGS richiede una biopsia dell'embrione per eseguire il test genetico. In alcune circostanze, i pazienti hanno un alto rischio di avere embrioni alterati, come i pazienti di età materna avanzata. In questi casi, c'è la possibilità che, dopo l'analisi PGS, tutti gli embrioni siano cromosomicamente anormali e non possano essere trasferiti.

Concetti Chiave in Genetica
Il mondo della genetica ha il suo gergo e le sue parole chiave.
Gene: Unità fisica di base dell’ereditarietà. È una sequenza di Dna che contiene l’informazione per la costruzione di una proteina o, in alcuni casi, di più proteine. Alcuni geni non codificano per proteine ma per molecole di Rna con particolari funzioni, per esempio funzioni di regolazione dell’espressione di altri geni. I geni possono avere lunghezze molto diverse: da poche centinaia a diverse migliaia di basi (le ‘lettere’ che compongono il Dna). Fisicamente, i geni sono posizionati in strutture chiamate cromosomi. Il Dna degli esseri umani contiene all’incirca 22 mila geni. Una piccolissima manciata di geni, inoltre, si trova nel Dna presente all’interno degli organuli che costituiscono le centrali energetiche delle cellule: i mitocondri.
Cellula: L'unità fondamentale della vita.
Autosomi: Cromosomi non sessuali, presenti in 22 coppie negli esseri umani. L'essere umano nasce con 46 cromosomi, 23 ereditati dal padre e 23 dalla madre e che trasmetteranno la nostra eredità. 22 sono cromosomi autosomi e 2 sono cromosomi sessuali, che distingueranno il sesso (XX nel caso delle donne e XY nel caso degli uomini).
Cromosoma: Una volta formata da molecole di Dna associate a proteine. Si divide per dare origine a due cellule figlie.
Alleli: Forme alternative di un gene o di una sequenza di Dna. L'uomo Gregor Mendel è stato il primo a stabilire le sue leggi dell’ereditarietà. Per un gene possono esistere più alleli, ad esempio uno codificante per il colore giallo e uno codificante per il colore verde. Gli alleli sono numerosi.
Ereditarietà: La trasmissione di tratti da genitore a figlio.
Dominante: Un allele dominante si esprime sempre quando è presente.
Recessivo: Un allele recessivo deve essere presente in duplice copia per potersi esprimere. Un allele dominante è in grado di mascherare l'espressione di un allele recessivo, ovvero è l’allele dominante a esprimersi.
Mutazione: Cambiamenti nella sequenza di lettere del Dna. Le mutazioni possono avere origini differenti: in genere sono causate da ‘semplici’ errori durante la fase di duplicazione del Dna che precede la divisione cellulare. In pratica: per dividersi dando origine a due cellule figlie, una cellula deve prima duplicare il proprio Dna, in modo da passarne una copia completa a ciascuna cellula figlia. Durante questa operazione, però, può capitare qualche errore, esattamente come accade quando si copia un testo. Questo errore è una mutazione. Altre mutazioni possono essere provocate dall’esposizione a certi agenti fisici, come le radiazioni ionizzanti, o chimici. Le mutazioni che interessano le cellule sessuali (ovociti e spermatozoi) vengono passate alla generazione successiva, mentre quelle che riguardano le altre cellule del corpo non possono essere passate ai figli. A volte questi cambiamenti riguardano sequenze estese di Dna, con intere porzioni di cromosomi che vengono perse o scambiate di posto. Possono anche riguardare cromosomi interi, come accade nel caso della sindrome di Down o trisomia 21, caratterizzata dalla presenza di una copia in più del cromosoma 21.
Omozigote: Un individuo che per un determinato gene, ha ereditato dai genitori due copie identiche di alleli.
Genotipo: L’insieme delle caratteristiche genetiche di un individuo. È la particolare composizione di alleli ereditati per un determinato gene. Le informazioni genetiche di un individuo sono chiamate genotipo.
Fenotipo: L’insieme delle caratteristiche osservabili di un individuo (altezza, colore degli occhi, ecc.).
Genoma: Set completo delle istruzioni genetiche che si trovano in una cellula. Negli esseri umani, per genoma si intende quello contenuto nel nucleo delle cellule, costituito da circa 3 miliardi di lettere di Dna, organizzate in 23 coppie di cromosomi. Oltre al genoma nucleare, nelle nostre cellule c’è anche un piccolo genoma mitocondriale, contenuto negli organuli che costituiscono le centrali energetiche cellulari. Rappresenta l’1% circa del genoma. Le informazioni geniche non sono del tutto chiare.

tags: #anticipazione #materna #genetica