L’anemia mediterranea, nota scientificamente come talassemia, rappresenta un complesso gruppo di malattie ereditarie che influenzano la sintesi dell’emoglobina, la proteina contenuta nei globuli rossi deputata al trasporto dell’ossigeno dai polmoni ai tessuti. Questa patologia, ampiamente diffusa nel bacino del Mediterraneo, in Italia, Grecia e altre regioni, è stata osservata per la prima volta proprio nelle popolazioni di quest’area, guadagnandosi il nome di "anemia mediterranea".
Basi genetiche e meccanismi della talassemia
Anche il sangue può essere interessato da malattie genetiche. Una, in particolare, riguarda da vicino gli italiani, e in genere le popolazione del bacino del Mediterraneo. Si tratta della talassemia o anemia mediterranea, chiamata così proprio perché osservate per la prima volta nelle popolazioni di questa area. L’anemia mediterranea è una malattia ereditaria, che viene trasmessa dai genitori ai figli al momento del concepimento. La trasmissione è dunque autosomico recessiva.
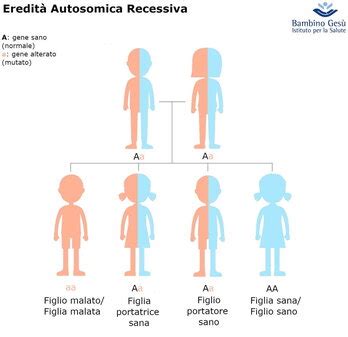
Le talassemie rappresentano un gruppo di malattie genetiche dovute ad anomalie della struttura dell’emoglobina (Hb). Dal punto di vista chimico, la molecola dell’emoglobina è costituita da 4 “globine”, proteine associate ciascuna a un gruppo non proteico contenente ferro e denominato “eme”. Da questa unione deriva il nome “emo-globina”. Quello che accade nella talassemia è che i geni dai quali dipende la produzione di queste catene non funzionano. Il risultato di questa anomalia dell’emoglobina è che i globuli rossi tendono a distruggersi precocemente. L’anemia mediterranea riduce la produzione di emoglobina ed aumenta il tasso di distruzione dei globuli rossi nel sangue, causando così anemia.
Classificazione delle talassemie: Alfa e Beta
Esistono più talassemie. In base al tipo (α, β, δβ, γδβ, δ) di catena globinica prodotta in difetto, si distinguono talassemie ed emoglobinopatie talassemiche. La beta-talassemia è circa 5 volte più frequente rispetto alla alfa-talassemia. Le β-talassaemie sono molto eterogenee dal punto di vista molecolare. Finora sono stati definiti oltre cento difetti, per lo più dovuti a singola sostituzione nucleotidica e raramente a grossolana delezione del gene β−globinico. Le α−talassemie sono dovute alla produzione difettosa (α+−talassemia) o totalmente assente (α0−talassemia) di catene globiniche.
Le forme minori (eterozigoti)
Il portatore di microcitemia è una persona sana, e può ignorare anche per tutta la vita, se non fa l’esame specifico, di avere nel sangue questa anomalia ereditaria. Nella beta-talassemia eterozigote o talassemia minore, nella quale i soggetti sono portatori della malattia, la sintomatologia clinica è negativa. Può tutt’al più avere un colorito un po’ pallido ed essere talvolta un po’ fiacco.
Le forme maggiori e intermedie
La forma omozigote della b-talassemia, invece, tende a manifestarsi nella primissima infanzia (entro 1-2 anni). La beta-talassemia omozigote o talassemia maggiore o morbo di Cooley è la forma più grave della malattia che si manifesta nei primi mesi di vita con un’anemia emolitica cronica. Esistono anche forme di talassemia omozigote meno gravi, spesso definite talassemie intermedie.
Segni clinici e sintomi
Subito dopo la nascita, ai test di laboratorio si rivela un'anemia che tende rapidamente ad aggravarsi. Nel giro del primo-secondo anno di età il bambino affetto dalla malattia si presenta pallido, itterico e la sua crescita è inferiore alla norma. Altri segni importanti sono le deformazioni ossee, dovute al fatto che il midollo si accresce in modo anomalo per compensare il difetto nella produzione di emoglobina, e l'ingrossamento di fegato e milza (epatosplenomegalia). Nei casi più gravi si presenta anche scompenso cardiaco dovuto alla dilatazione del cuore.
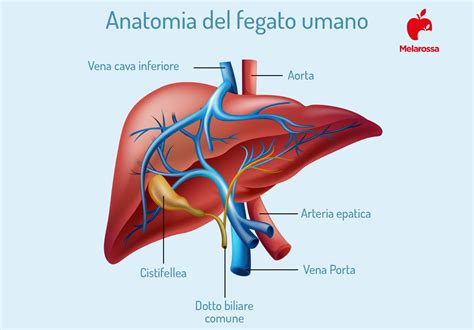
Se la talassemia maior non viene trattata, l'ingrossamento di fegato e milza aumenta, le ossa divengono pericolosamente fragili e, soprattutto, si ha la dilatazione del muscolo cardiaco. Il danno a carico di fegato e milza compromettono tra l'altro le difese immunitarie, ragion per cui le malattie infettive e l'insufficienza cardiaca sono le principali cause di morte tra i malati non trattati.
Diagnosi e screening
La diagnosi di anemia mediterranea si basa oggi su un protocollo clinico consolidato che combina l’analisi della storia familiare con esami di laboratorio di precisione. Il primo sospetto nasce solitamente da un normale emocromo. Nei soggetti con talassemia, si osserva tipicamente una microcitosi, ovvero globuli rossi di dimensioni ridotte (basso valore di MCV), associata a una riduzione dei livelli di emoglobina.
Il "gold standard" per la diagnosi è l’elettroforesi dell’emoglobina o, più comunemente oggi, la cromatografia liquida ad alta prestazione (HPLC). Questi test permettono di quantificare i diversi tipi di emoglobina presenti (come l’emoglobina A2 e l’emoglobina F). L’analisi del DNA (test molecolari) rappresenta lo step definitivo. Viene utilizzata per identificare le specifiche mutazioni genetiche responsabili della malattia.
Terapia trasfusionale e gestione del ferro
Nella maggior parte delle persone con talassemia major o altre forme gravi è necessario effettuare regolari trasfusioni di sangue per curare l’anemia. La frequenza con cui devono essere effettuate dipende dal tipo di talassemia da cui si è colpiti. Se si tratta della forma più grave, la beta-talassemia major, può essere necessaria una trasfusione di sangue una volta al mese.
Trasfusioni molto frequenti possono causare accumulo di ferro nel corpo e richiedere medicinali specifici per la sua eliminazione. La cura per rimuovere il ferro in eccesso causato da regolari trasfusioni di sangue è nota come terapia di chelazione e va iniziata dopo circa 10 trasfusioni. I medicinali usati nella terapia di chelazione sono conosciuti come agenti chelanti.
Terapia ferrochelante, Angelucci: "Cagliari in prima linea"
Per valutare il carico di ferro, si utilizzano diverse metodiche:
- LIC: test che misura la concentrazione del ferro nel fegato.
- SQUID: nuova metodica non invasiva utilizzata per valutare accuratamente l’eccesso di ferro. Si basa sulle proprietà paramagnetiche del ferro. È una tecnica purtroppo molto costosa.
- RMN: risonanza magnetica nucleare per immagini, un metodo non invasivo e più facilmente disponibile sul territorio nazionale, per misurare l’accumulo di ferro nel fegato.
Opzioni terapeutiche avanzate e trapianto
L’unica cura (terapia) definitiva contro l’anemia mediterranea è il trapianto di midollo osseo. Esso consiste nella somministrazione di cellule staminali, prelevate dal midollo osseo di un donatore compatibile, che andranno a sostituire quelle difettose del malato. Il trapianto di cellule staminali è una cura intensiva che comporta una serie di rischi come, ad esempio, il rigetto delle cellule trapiantate o la malattia del trapianto contro l’ospite.
In questi ultimi anni sono stati fatti notevoli progressi nella sperimentazione di terapie alternative al trapianto di midollo. La terapia genica è oggi una potenziale opzione di trattamento quando manca un donatore compatibile. La prima forma consiste nell’inserire nelle cellule staminali del paziente il gene da cui dipende la produzione della b-globina. La seconda forma mira a indurre l’organismo a continuare a produrre emoglobina fetale per tutta la vita.
Fertilità e salute riproduttiva
Per fertilità s’intende la capacità di avere e portare a termine una gravidanza: una ridotta fertilità è comune agli individui affetti da anemia mediterranea dipendente da trasfusioni, come la beta-talassemia maggiore. La fertilità può essere ridotta a causa di un eccesso di ferro nella ghiandola pituitaria (ipofisi): il danno che ne deriva impedisce il rilascio di ormoni ipofisari in risposta ai segnali provenienti dall’ipotalamo.
Sembra che l’approccio migliore per cercare una gravidanza quando si ha l’anemia mediterranea consista nel tenere sotto controllo i livelli di ferro. I pazienti che agiscono al meglio sono quelli che cominciano precocemente il trattamento, prima che i livelli di ferro, misurati in termini di incremento del livello di ferritina, diventino troppo elevati.
Anomalie congenite e monitoraggio fetale
Durante le fasi di sviluppo fetale può verificarsi l’insorgenza di malformazioni congenite anche definite anomalie congenite. Ad esempio, la misurazione della translucenza nucale, eseguibile tra la 11a e la 13a settimana gestazionale permette, in combinazione con il dosaggio ematico di alcuni marcatori biochimici, di valutare il rischio di alcune cromosomopatie.
Nel secondo trimestre di gestazione la valutazione ecografica permette l’identificazione di molte malformazioni congenite attraverso lo studio accurato dell’anatomia fetale. La diagnosi prenatale non invasiva eseguita su sangue materno analizza il rischio delle trisomie fetali più comuni e aneuploidie X, Y in gravidanze dalla decima settimana in poi. L’esame ha dimostrato un’attendibilità superiore al 99% nel determinare il rischio di trisomia 21. Le infezioni in gravidanza rappresentano una causa biologica rilevante nell’eziologia di alcune malformazioni fetali. La consulenza teratologica per esposizione a farmaci in gravidanza mira a valutare i possibili rischi fetali in relazione al tipo, alla durata e all’epoca di esposizione.
tags: #anomalia #fetale #mediterraneo